
Overview
Beisi (pronounce /bās/ as bass) is a highly motivated, skilled, communicative, independent and team-oriented PhD biologist with over 10 years of bioinformatics experience (who did not know how to play bass). He has extensive expertise in Epigenomics, Cancer Biology, Developmental Biology, and High Performance Computation.
NGS Data Beisi knows how to analyze:
- CHIP-Seq
- ATAC-Seq
- RNA-Seq
- Hi-C(Juicer)
- HiCHIP/PLAC-seq/Capture-C(HiC-Pro)
- scRNA-Seq(Seurat/Pagoda)
- scATAC-Seq
- SLAM-Seq(SlamDunk)
- DNase-Seq, FAIRE-Seq, MNase-Seq
- WGBS/TEBS
Education
- PhD, University of Science and Technology of China, P.R.CHINA (2009)
- Research Fellowship, Indiana University-Purdue University at Indianapolis, USA (2007)
- BS, University of Science and Technology of China, P.R.CHINA (2003)
Professional Experience
| Time | Position | PI/Supervisor | Institution |
|---|---|---|---|
| 2021- | Principal Bioinformatics | Gang Wu / | St. Jude Children’s Research Hospital, Memphis, TN |
| Research Scientist | Yiping Fan | ||
| 2019-2021 | Group Lead in Epigenetics | Gang Wu / | St. Jude |
| Yiping Fan | |||
| 2017-2019 | Sr. Bioinformatics | Jinghui Zhang / | St. Jude |
| Research Scientist | Yiping Fan | ||
| 2014-2017 | Bioinformatics | Jinghui Zhang / | St. Jude |
| Research Scientist | Yiping Fan | ||
| 2011-2014 | Postdoctoral Fellow | Dustin E.Schones | City of Hope, Duarte, CA |
| 2009-2011 | Postdoctoral Fellow | Guohui Li | Dalian Institute of Chemical Physics, Dalian |
| 2009-2009 | Postdoctoral Fellow(Joint) | Yan Cui | University of Tennessee Health Science Center, Memphis, TN |
| 2009-2009 | Internship | BGI | Beijing Genomics Institute, Shenzhen |
| 2007-2008 | Research Fellow | Yaoqi Zhou | IUPUI, Indianapolis, IN |
| 2003-2007 | Research Assistant | Haojun Liang | University of Science and Technology of China, Hefei |
Publications
For a full list (GEO|SRA|Browser|Code) see below , or Google Scholar , or Pubmed
Featured
-
Deciphering the Role of RNA in Regulating CTCF’s DNA Binding Affinity in Leukemia Cells 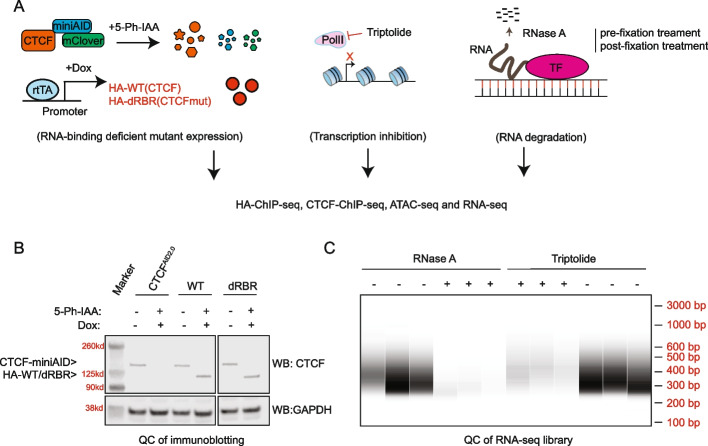
CTCF, a highly studied transcription factor, is essential for chromatin interaction maintenance. Several independent studies report that CTCF interacts with RNAs in vitro and in cells. Several independent studies report that CTCF interacts with RNAs in vitro and in cells. Yet continuous debates about the authenticity of the RNA-binding affinity of CTCF and its biological role remain in large part due to limited research techniques available, such as CLIP-seq
-
CTCF Is Selectively Required for Maintaining Chromatin Accessibility and Gene Expression in Human Erythropoiesis 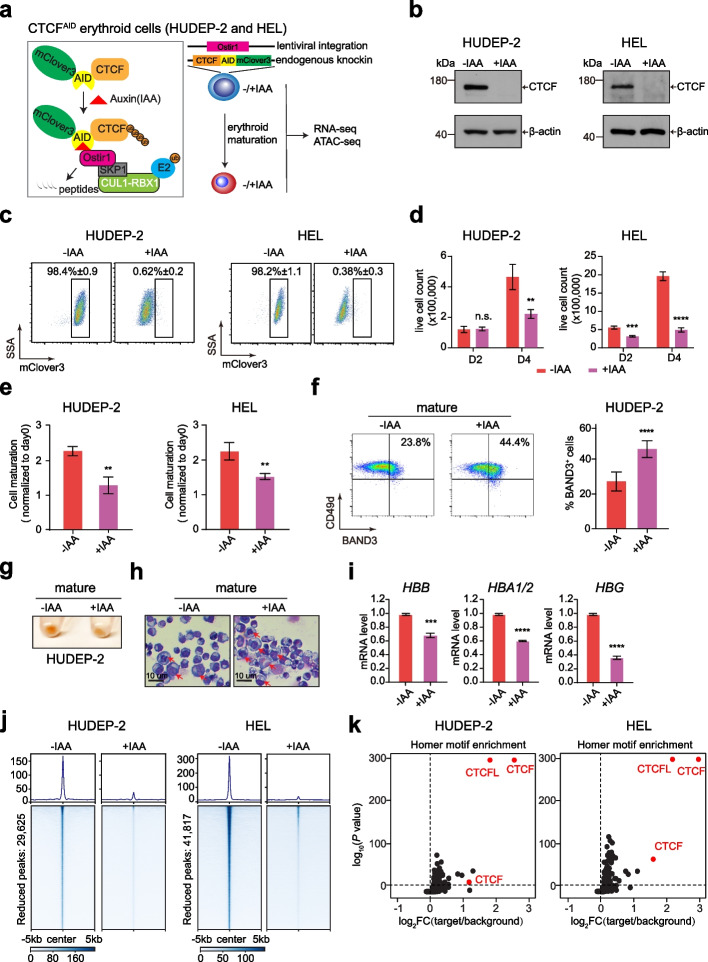
CTCF is considered as the most essential transcription factor regulating chromatin architecture and gene expression. However, genome-wide impact of CTCF on erythropoiesis has not been extensively investigated. CTCF is considered as the most essential transcription factor regulating chromatin architecture and gene expression. However, genome-wide impact of CTCF on erythropoiesis has not been extensively investigated
-
Dynamic Foxp3–Chromatin Interaction Controls Tunable Treg Cell Function 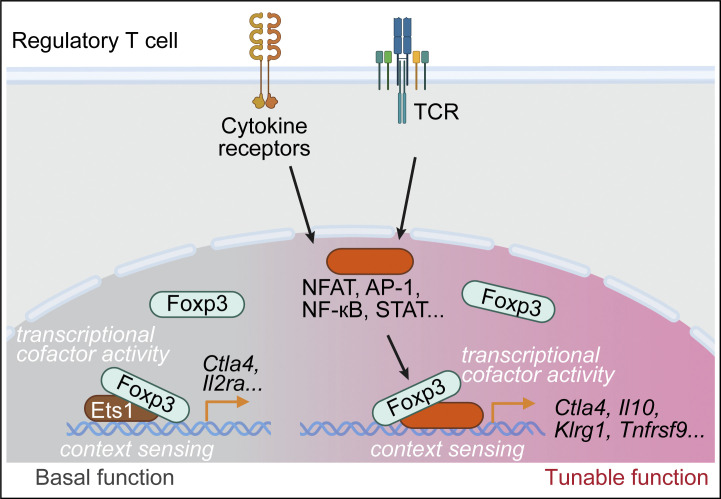
Nuclear factor Foxp3 determines regulatory T (Treg) cell fate and function via mechanisms that remain unclear. Here, we investigate the nature of Foxp3-mediated gene regulation in suppressing autoimmunity and antitumor immune response. Furthermore, mutations at the Foxp3 DNA-binding domain destabilize the Foxp3–chromatin association. These representative settings delineate context-dependent Foxp3–chromatin interaction, suggesting that Foxp3 associates with chromatin by hijacking DNA-binding proteins resulting from Treg activation or differentiation, which is stabilized by direct Foxp3–DNA binding, to dynamically regulate Treg cell function according to immunological contexts
-
THUMPD2 Catalyzes the N2-methylation of U6 snRNA of the Spliceosome Catalytic Center and Regulates Pre-mRNA Splicing and Retinal Degeneration 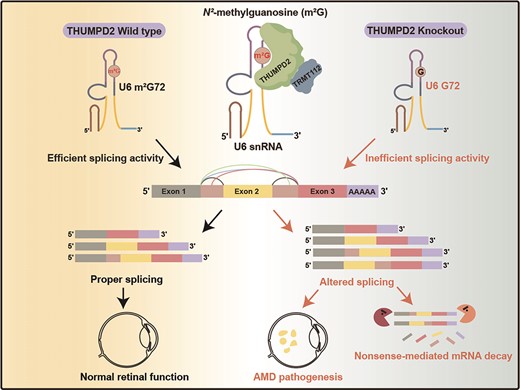
The mechanisms by which the relatively conserved spliceosome manages the enormously large number of splicing events that occur in humans (∼200 000 versus ∼300 in yeast) are poorly understood. Here, we show deposition of one RNA modification-N2-methylguanosine (m2G) on the G72 of U6 snRNA (the catalytic center of the spliceosome) promotes efficient pre-mRNA splicing activity in human cells. We further show that THUMPD2 was associated with age-related macular degeneration and retinal function. Our study thus demonstrates how an RNA epigenetic modification of the major spliceosome regulates global pre-mRNA splicing and impacts physiology and disease
-
PP2Ac Deficiency Enhances Tumor Immunogenicity by Activating STING-Type I Interferon Signaling in Glioblastoma 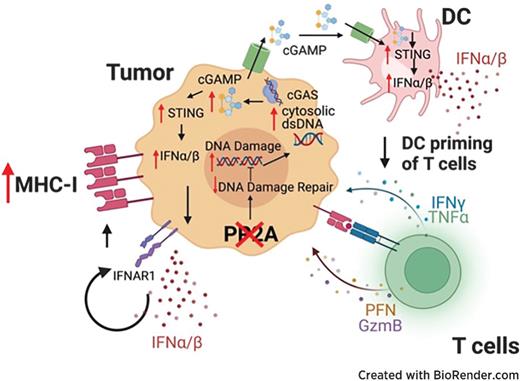
Glioblastoma (GBM) is an immunologically “cold” tumor that does not respond to current immunotherapy. Here, we demonstrate a fundamental role for the α-isoform of the catalytic subunit of protein phosphatase-2A (PP2Ac) in regulating glioma immunogenicity. Furthermore, loss of PP2Ac increased IFN signaling in myeloid and tumor cells and reduced expression of a tumor gene signature associated with worse patient survival in TCGA. Collectively, this study establishes a novel role for PP2Ac in inhibiting dsDNA-cGAS-STING signaling to suppress anti-tumor immunity in glioma
-
PP2Ac/STRN4 Negatively Regulates STING-Type I Interferon Signaling in Tumor Associated Macrophages 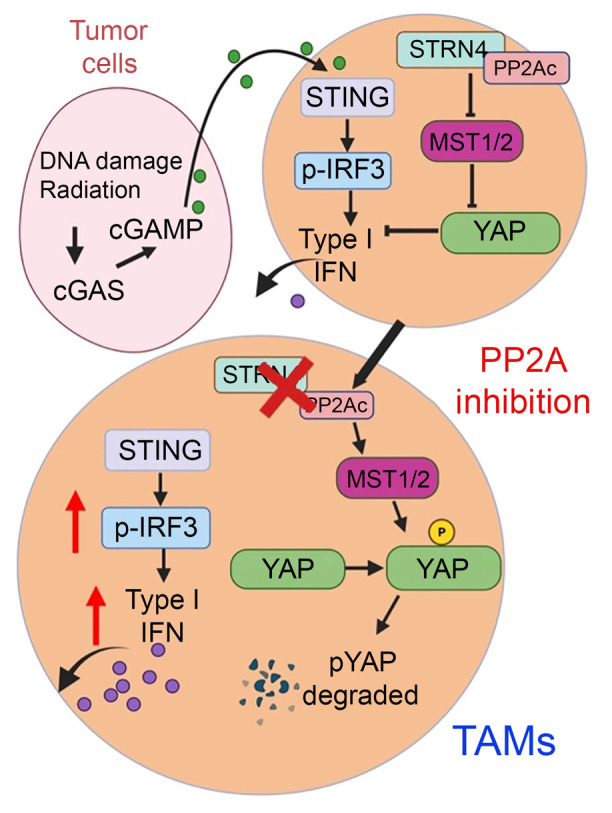
Stimulator of IFN genes type I (STING-Type I) IFN signaling in myeloid cells plays a critical role in effective antitumor immune responses, but STING agonists as monotherapy have shown limited efficacy in clinical trials. The mechanisms that downregulate STING signaling are not fully understood. We also demonstrated that PP2A/STRN4 deficiency in macrophages reduced YAP/TAZ expression and sensitized tumor-conditioned macrophages to STING stimulation. In summary, we demonstrated that PP2A/STRN4-YAP/TAZ has, in our opinion, been an unappreciated mechanism that mediates immunosuppression in tumor-associated macrophages, and targeting the PP2A/STRN4-YAP/TAZ axis can sensitize tumors to immunotherapy
-
Auxin-Inducible Degron 2 System Deciphers Functions of CTCF Domains in Transcriptional Regulation 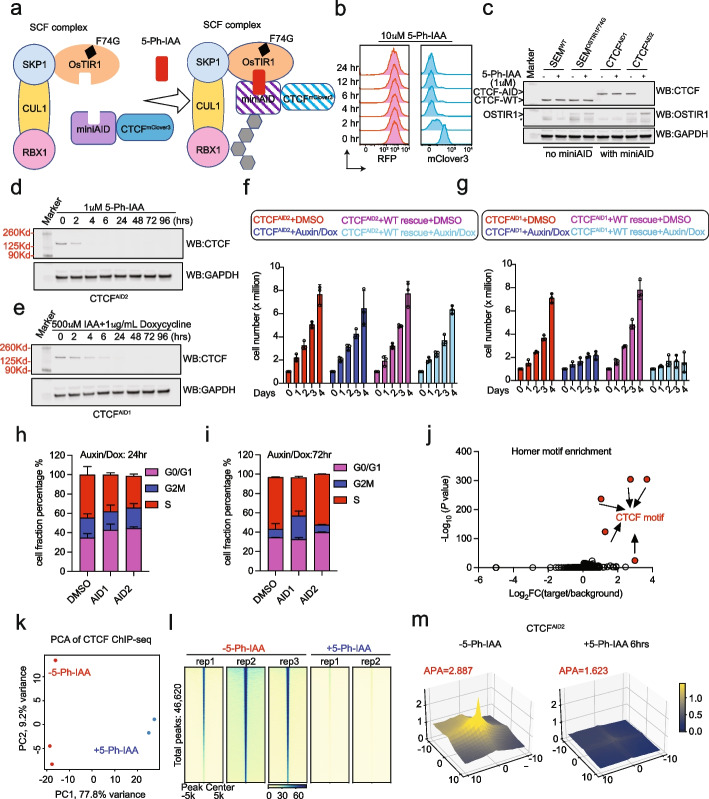
CTCF is a well-established chromatin architectural protein that also plays various roles in transcriptional regulation. While CTCF biology has been extensively studied, how the domains of CTCF function to regulate transcription remains unknown. While CTCF biology has been extensively studied, how the domains of CTCF function to regulate transcription remains unknown. Additionally, the original auxin-inducible degron 1 (AID1) system has limitations in investigating the function of CTCF
-
Genomic Profiling Identifies Genes and Pathways Dysregulated by HEY1-NCOA2 Fusion and Shines a Light on Mesenchymal Chondrosarcoma Tumorigenesis 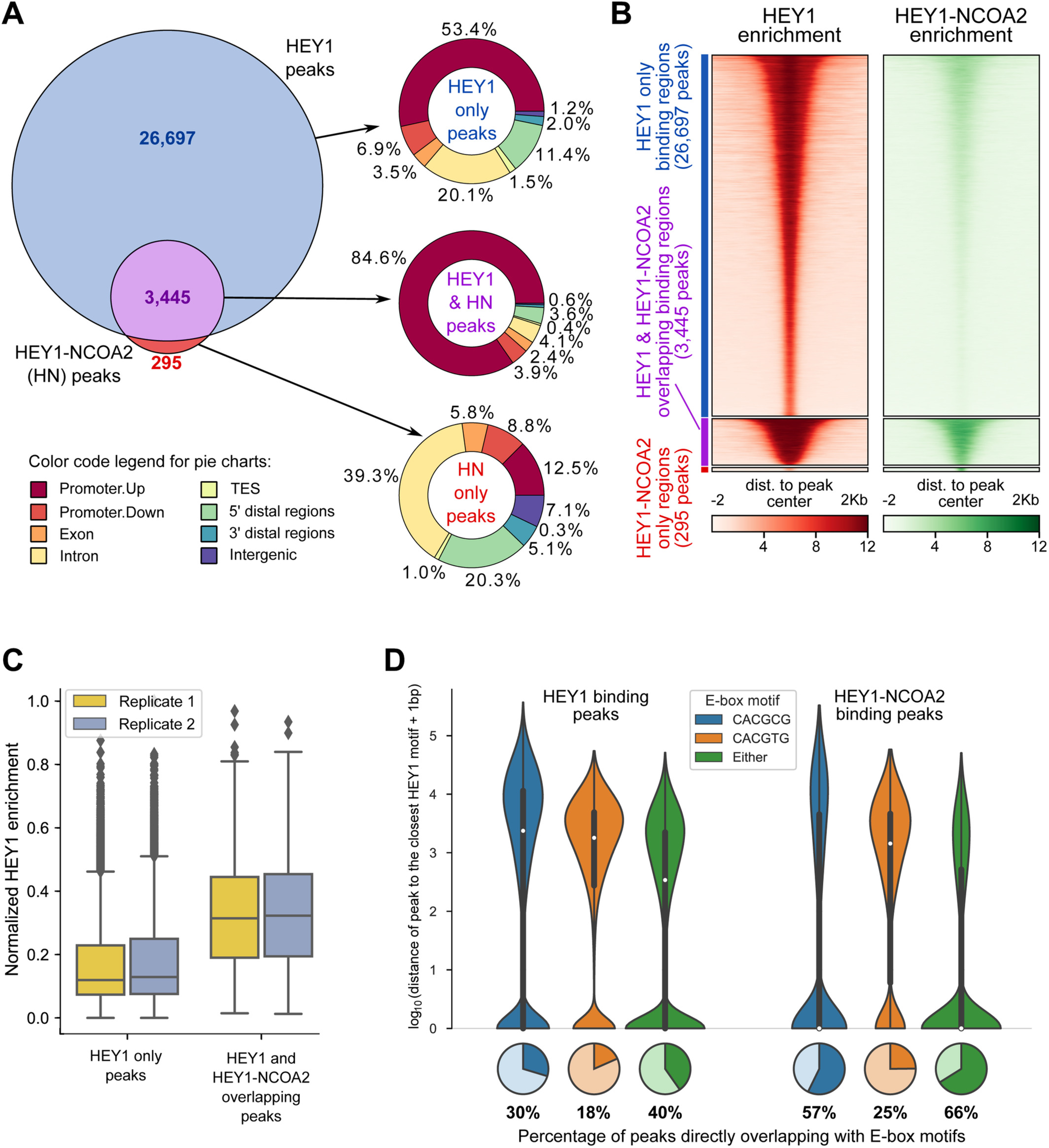
Mesenchymal chondrosarcoma is a rare, high-grade, primitive mesenchymal tumor. It accounts for around 2-10% of all chondrosarcomas and mainly affects adolescents and young adults. \textcopyright 2022 The Authors. The Journal of Pathology published by John Wiley & Sons Ltd on behalf of The Pathological Society of Great Britain and Ireland
-
A Dual Role of Human tRNA Methyltransferase hTrmt13 in Regulating Translation and Transcription 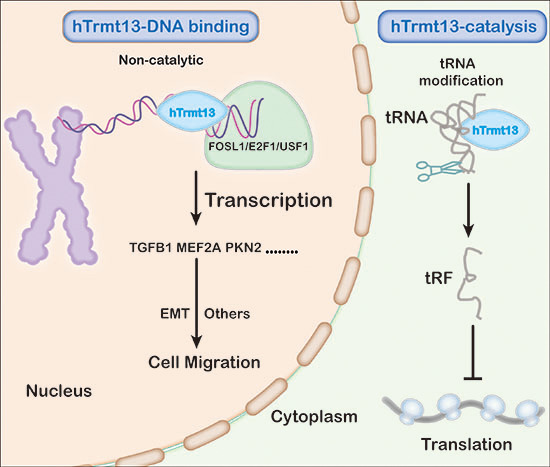
Since numerous RNAs and RBPs prevalently localize to active chromatin regions, many RNA-binding proteins (RBPs) may be potential transcriptional regulators. RBPs are generally thought to regulate transcription via noncoding RNAs. Finally, we find that hTrmt13 expression is tightly associated with poor prognosis and survival in diverse cancer patients. Our discovery of the noncatalytic roles of an RNA-modifying enzyme provides a new perspective for understanding epitranscriptomic regulation
-
Control of Foxp3 Induction and Maintenance by Sequential Histone Acetylation and DNA Demethylation 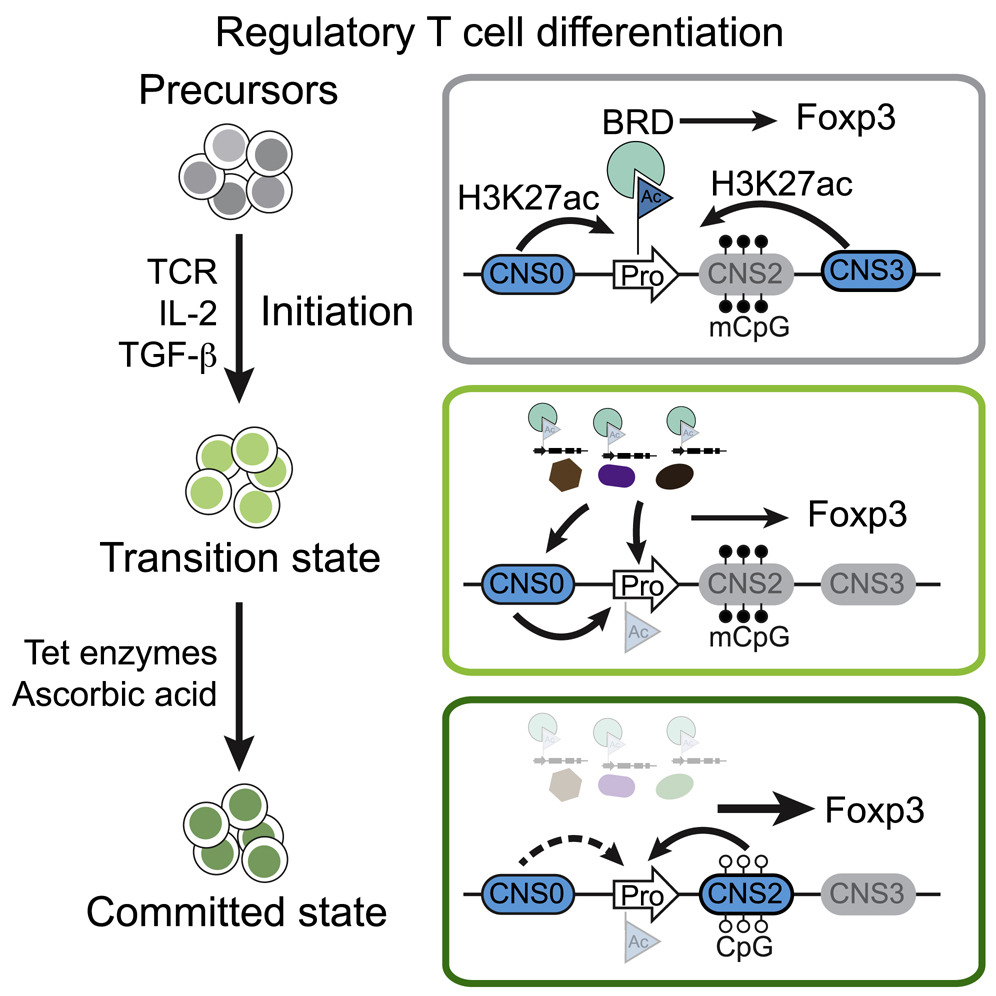
Regulatory T (Treg) cells play crucial roles in suppressing deleterious immune response. Here, we investigate how Treg cells are mechanistically induced in vitro (iTreg) and stabilized via transcriptional regulation of Treg lineage-specifying factor Foxp3. These processes transform stochastic iTreg induction into a stable cell fate, with the former sensitive and the latter resistant to genetic and environmental perturbations. Thus, sequential histone acetylation and DNA demethylation in Foxp3 induction and maintenance reflect stepwise mechanical switches governing iTreg cell lineage specification
-
Acute Depletion of CTCF Rewires Genome-Wide Chromatin Accessibility 
Background The transcription factor CTCF appears indispensable in defining topologically associated domain boundaries and maintaining chromatin loop structures within these domains, supported by numerous functional studies. However, acute depletion of CTCF globally reduces chromatin interactions but does not significantly alter transcription. Interestingly, many CTCF co-regulators that have alterations of their respective downstream gene expression do not show changes of their own expression levels across the multi-omics measurements upon acute CTCF loss, highlighting the strength of our system to discover hidden co-regulatory partners associated with CTCF-mediated transcription. Conclusions This study highlights that CTCF loss rewires genome-wide chromatin accessibility, which plays a critical role in transcriptional regulation
-
Ybx1 Fine-Tunes PRC2 Activities to Control Embryonic Brain Development 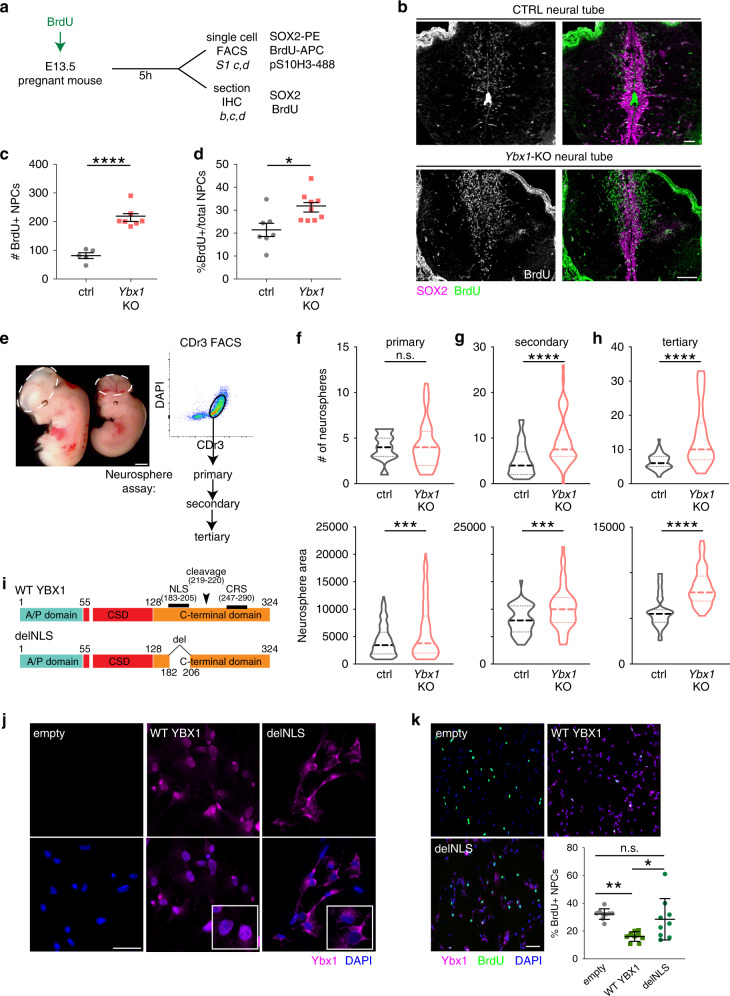
Chromatin modifiers affect spatiotemporal gene expression programs that underlie organismal development. The Polycomb repressive complex 2 (PRC2) is a crucial chromatin modifier in executing neurodevelopmental programs. In Ybx1-knockout NPCs, H3K27me3 reduction by PRC2 enzymatic inhibitor or genetic depletion partially rescues gene expression and NPC functions. Our findings suggest that Ybx1 fine-tunes PRC2 activities to regulate spatiotemporal gene expression in embryonic neural development and uncover a crucial epigenetic mechanism balancing forebrain–hindbrain lineages and self-renewal–differentiation choices in NPCs
-
Targeting REGNASE-1 Programs Long-Lived Effector T Cells for Cancer Therapy 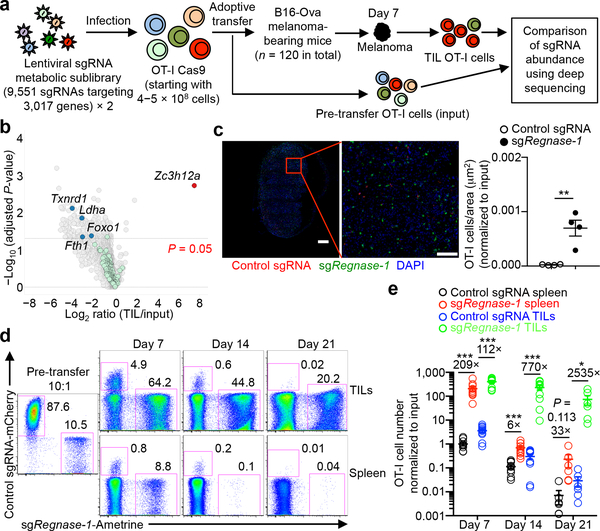
Adoptive cell therapy represents a new paradigm in cancer immunotherapy, but it can be limited by the poor persistence and function of transferred T cells1. Here we use an in vivo pooled CRISPR–Cas9 mutagenesis screening approach to demonstrate that, by targeting REGNASE-1, CD8+ T cells are reprogrammed to long-lived effector cells with extensive accumulation, better persistence and robust effector function in tumours. By contrast, the targeting of additional signalling factors—including PTPN2 and SOCS1—improves the therapeutic efficacy of REGNASE-1-deficient CD8+ T cells. Our findings suggest that T cell persistence and effector function can be coordinated in tumour immunity and point to avenues for improving the efficacy of adoptive cell therapy for cancer
-
Nucleome Dynamics during Retinal Development 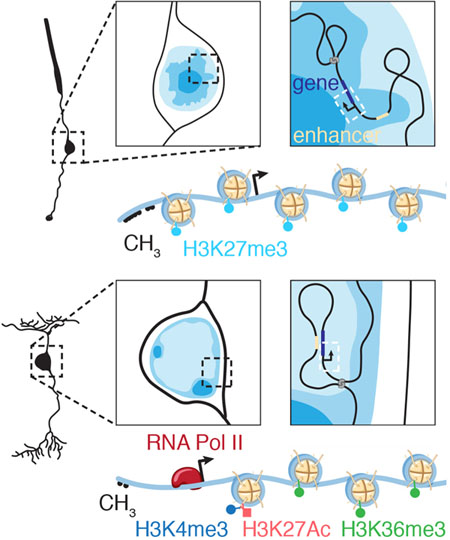
Summary More than 8,000 genes are turned on or off as progenitor cells produce the 7 classes of retinal cell types during development. Thousands of enhancers are also active in the developing retinae, many having features of cell- and developmental stage-specific activity. Single-cell ATAC-seq and RNA-seq were integrated with our Hi-C and previous ChIP-seq data to identify cell- and developmental-stage-specific super-enhancers (SEs). We identified a bipolar neuron-specific core regulatory circuit SE upstream of Vsx2, whose deletion in mice led to the loss of bipolar neurons
-
Acute Depletion of CTCF Directly Affects MYC Regulation through Loss of Enhancer–Promoter Looping 
Numerous pieces of evidence support the complex, 3D spatial organization of the genome dictates gene expression. CTCF is essential to define topologically associated domain boundaries and to facilitate the formation of insulated chromatin loop structures. 8 Mb downstream. Notably, MYC expression was not profoundly affected upon CTCF loss in HUDEP-2 cells suggesting that CTCF could play a B-ALL cell line specific role in maintaining MYC expression
-
Differentiation of Human Pluripotent Stem Cells into Neurons or Cortical Organoids Requires Transcriptional Co-Regulation by UTX and 53BP1 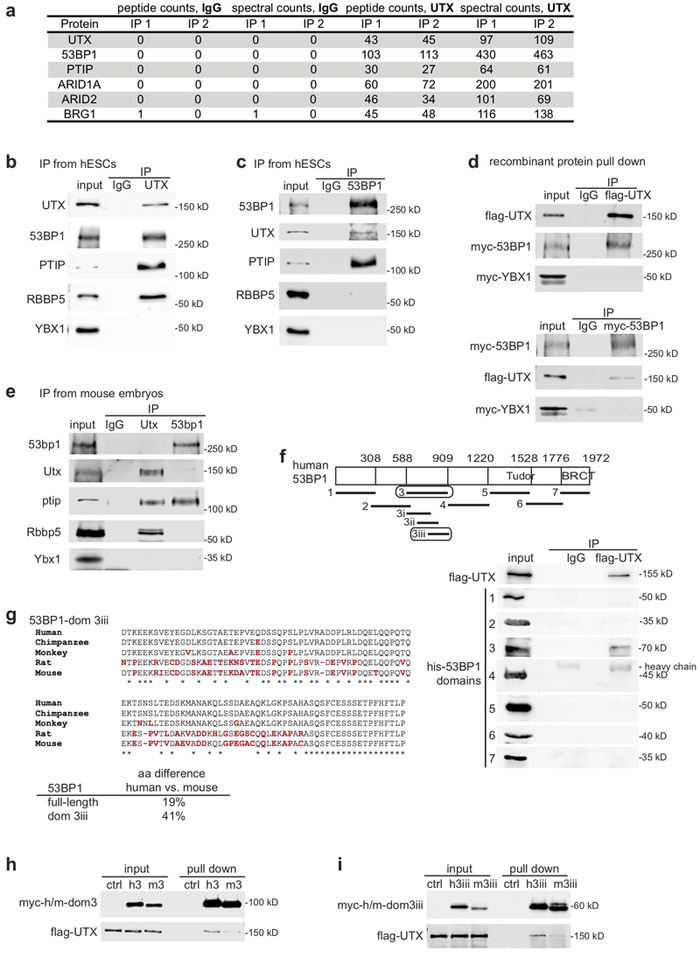
UTX is a chromatin modifier required for development and neural lineage specification, but how it controls these biological processes is unclear. To determine the molecular mechanisms of UTX, we identified novel UTX protein interaction partners. 53BP1 promotes UTX chromatin binding, and in turn H3K27 modifications and gene activation, at a subset of genomic regions, including neurogenic genes. Overall, our data suggest that the 53BP1–UTX interaction supports the activation of key genes required for human neurodevelopment
-
Metabolic Heterogeneity Underlies Reciprocal Fates of T H 17 Cell Stemness and Plasticity 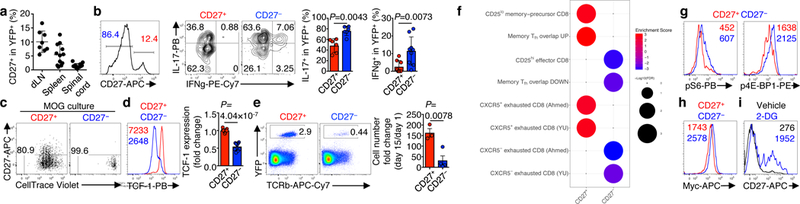
A defining feature of adaptive immunity is the development of long-lived memory T cells to curtail infection. Recent studies have identified a unique stem-like T-cell subset amongst exhausted CD8-positive T cells in chronic infection1–3, but it remains unclear whether CD4-positive T-cell subsets with similar features exist in chronic inflammatory conditions. Single-cell RNA sequencing and experimental validation reveal heterogeneity in fate-mapped TH17 cells, and a developmental arrest in the TH1 transdifferentiation trajectory upon loss of mTORC1 activity or metabolic perturbation. Our results establish that the dichotomy of stemness and effector function underlies the heterogeneous TH17 responses and autoimmune pathogenesis, and point to previously unappreciated metabolic control of plasticity in helper T cells
-
Retinal Cell Type DNA Methylation and Histone Modifications Predict Reprogramming Efficiency and Retinogenesis in 3D Organoid Cultures 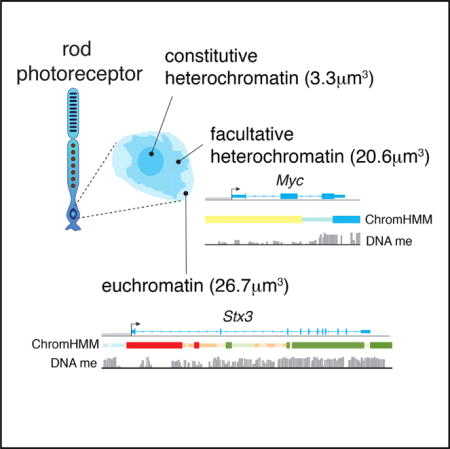
Summary Diverse cell types can be reprogrammed into pluripotent stem cells by ectopic expression of Oct4 (Pou5f1), Klf4, Sox2, and Myc. Many of these induced pluripotent stem cells (iPSCs) retain memory, in terms of DNA methylation and histone modifications (epigenetic memory), of their cellular origins, and this may bias subsequent differentiation. Neurons with the lowest reprogramming efficiency produced iPSC lines with the best retinal differentiation and were more likely to retain epigenetic memory of their cellular origins. In addition, we identified biomarkers of iPSCs that are predictive of retinal differentiation
-
The Dynamic Epigenetic Landscape of the Retina During Development, Reprogramming, and Tumorigenesis 
In the developing retina, multipotent neural progenitors undergo unidirectional differentiation in a precise spatiotemporal order. Here we profile the epigenetic and transcriptional changes that occur during retinogenesis in mice and humans. The retinoblastoma epigenome mapped to the developmental stage when retinal progenitors switch from neurogenic to terminal patterns of cell division. The epigenome of retinoblastomas was more similar to that of the normal retina than that of retina-derived iPSCs, and we identified retina-specific epigenetic memory
Full List
2025:3
-
Deciphering the Role of RNA in Regulating CTCF’s DNA Binding Affinity in Leukemia CellsGenome Biology 2025
CTCF, a highly studied transcription factor, is essential for chromatin interaction maintenance. Several independent studies report that CTCF interacts with RNAs in vitro and in cells. Yet continuous debates about the authenticity of the RNA-binding affinity of CTCF and its biological role remain in large part due to limited research techniques available, such as CLIP-seq.
-
CTCF Is Selectively Required for Maintaining Chromatin Accessibility and Gene Expression in Human ErythropoiesisGenome Biology 2025
CTCF is considered as the most essential transcription factor regulating chromatin architecture and gene expression. However, genome-wide impact of CTCF on erythropoiesis has not been extensively investigated.
-
Latent Epigenetic Programs in Müller Glia Contribute to Stress and Disease Response in the RetinaDevelopmental Cell 2025
2024:11
-
Phosphorylation of the DNA Damage Repair Factor 53BP1 by ATM Kinase Controls Neurodevelopmental Programs in Cortical Brain OrganoidsPLOS Biology 2024
53BP1 is a well-established DNA damage repair factor that has recently emerged to critically regulate gene expression for tumor suppression and neural development. However, its precise function and regulatory mechanisms remain unclear. Here, we showed that phosphorylation of 53BP1 at serine 25 by ATM is required for neural progenitor cell proliferation and neuronal differentiation in cortical brain organoids. Dynamic phosphorylation of 53BP1-serine 25 controls 53BP1 target genes governing neuronal differentiation and function, cellular response to stress, and apoptosis. Mechanistically, ATM and RNF168 govern 53BP1’s binding to gene loci to directly affect gene regulation, especially at genes for neuronal differentiation and maturation. 53BP1 serine 25 phosphorylation effectively impedes its binding to bivalent or H3K27me3-occupied promoters, especially at genes regulating H3K4 methylation, neuronal functions, and cell proliferation. Beyond 53BP1, ATM-dependent phosphorylation displays wide-ranging effects, regulating factors in neuronal differentiation, cytoskeleton, p53 regulation, as well as key signaling pathways such as ATM, BDNF, and WNT during cortical organoid differentiation. Together, our data suggest that the interplay between 53BP1 and ATM orchestrates essential genetic programs for cell morphogenesis, tissue organization, and developmental pathways crucial for human cortical development.
-
PHF6 Cooperates with SWI/SNF Complexes to Facilitate Transcriptional ProgressionNature Communications 2024
Here, the authors identify PHF6 as a dependency in SMARCB1-deficient rhabdoid cancers. Mechanistically, this study suggests a regulatory role for PHF6 in recruiting SWI/SNF complexes to enable RNA polymerase progression.
-
Evolutionary Conservation of VSX2 Super-Enhancer Modules in Retinal DevelopmentDevelopment 2024
Super-enhancers (SEs) are expansive regions of genomic DNA that regulate the expression of genes involved in cell identity and cell fate. We recently identified developmental stage- and cell type-specific modules within the murine Vsx2 SE. Here, we show that the human VSX2 SE modules have similar developmental stage- and cell type-specific activity in reporter gene assays. By inserting the human sequence of one VSX2 SE module into a mouse with microphthalmia, eye size was rescued. To understand the function of these SE modules during human retinal development, we deleted individual modules in human embryonic stem cells and generated retinal organoids. Deleting one module results in small organoids, recapitulating the small-eyed phenotype of mice with microphthalmia, while deletion of the other module led to disruptions in bipolar neuron development. This prototypical SE serves as a model for understanding developmental stage- and cell type-specific effects of neurogenic transcription factors with complex expression patterns. Moreover, by elucidating the gene regulatory mechanisms, we can begin to examine how dysregulation of these mechanisms contributes to phenotypic diversity and disease.
-
The TERT Promoter Is Polycomb-Repressed in Neuroblastoma Cells with Long TelomeresCancer Research Communications 2024
Acquiring a telomere maintenance mechanism is a hallmark of high-risk neuroblastoma and commonly occurs by expressing telomerase (TERT). Telomerase-negative neuroblastoma has long telomeres and utilizes the telomerase-independent alternative lengthening of telomeres (ALT) mechanism. Conversely, no discernable telomere maintenance mechanism is detected in a fraction of neuroblastoma with long telomeres. Here, we show, unlike most cancers, DNA of the TERT promoter is broadly hypomethylated in neuroblastoma. In telomerase-positive neuroblastoma cells, the hypomethylated DNA promoter is approximately 1.5 kb. The TERT locus shows active chromatin marks with low enrichment for the repressive mark, H3K27me3. MYCN, a commonly amplified oncogene in neuroblstoma, binds to the promoter and induces TERT expression. Strikingly, in neuroblastoma with long telomeres, the hypomethylated region spans the entire TERT locus, including multiple nearby genes with enrichment for the repressive H3K27me3 chromatin mark. Furthermore, subtelomeric regions showed enrichment of repressive chromatin marks in neuroblastomas with long telomeres relative to those with short telomeres. These repressive marks were even more evident at the genic loci, suggesting a telomere position effect (TPE). Inhibiting H3K27 methylation by three different EZH2 inhibitors induced the expression of TERT in cell lines with long telomeres and H3K27me3 marks in the promoter region. EZH2 inhibition facilitated MYCN binding to the TERT promoter in neuroblastoma cells with long telomeres. Taken together, these data suggest that epigenetic regulation of TERT expression differs in neuroblastoma depending on the telomere maintenance status, and H3K27 methylation is important in repressing TERT expression in neuroblastoma with long telomeres.The epigenetic landscape of the TERT locus is unique in neuroblastoma. The DNA at the TERT locus, unlike other cancer cells and similar to normal cells, are hypomethylated in telomerase-positive neuroblastoma cells. The TERT locus is repressed by polycomb repressive complex-2 complex in neuroblastoma cells that have long telomeres and do not express TERT. Long telomeres in neuroblastoma cells are also associated with repressive chromatin states at the chromosomal termini, suggesting TPE.
-
Dynamic Foxp3–Chromatin Interaction Controls Tunable Treg Cell FunctionJournal of Experimental Medicine 2024
Nuclear factor Foxp3 determines regulatory T (Treg) cell fate and function via mechanisms that remain unclear. Here, we investigate the nature of Foxp3-mediated gene regulation in suppressing autoimmunity and antitumor immune response. Contrasting with previous models, we find that Foxp3–chromatin binding is regulated by Treg activation states, tumor microenvironment, and antigen and cytokine stimulations. Proteomics studies uncover dynamic proteins within Foxp3 proximity upon TCR or IL-2 receptor signaling in vitro, reflecting intricate interactions among Foxp3, signal transducers, and chromatin. Pharmacological inhibition and genetic knockdown experiments indicate that NFAT and AP-1 protein Batf are required for enhanced Foxp3–chromatin binding in activated Treg cells and tumor-infiltrating Treg cells to modulate target gene expression. Furthermore, mutations at the Foxp3 DNA-binding domain destabilize the Foxp3–chromatin association. These representative settings delineate context-dependent Foxp3–chromatin interaction, suggesting that Foxp3 associates with chromatin by hijacking DNA-binding proteins resulting from Treg activation or differentiation, which is stabilized by direct Foxp3–DNA binding, to dynamically regulate Treg cell function according to immunological contexts.
-
A Single-Vector Intersectional AAV Strategy for Interrogating Cellular Diversity and Brain FunctionNature Neuroscience 2024
As discovery of cellular diversity in the brain accelerates, so does the need for tools that target cells based on multiple features. Here we developed Conditional Viral Expression by Ribozyme Guided Degradation (ConVERGD), an adeno-associated virus-based, single-construct, intersectional targeting strategy that combines a self-cleaving ribozyme with traditional FLEx switches to deliver molecular cargo to specific neuronal subtypes. ConVERGD offers benefits over existing intersectional expression platforms, such as expanded intersectional targeting with up to five recombinase-based features, accommodation of larger and more complex payloads and a vector that is easy to modify for rapid toolkit expansion. In the present report we employed ConVERGD to characterize an unexplored subpopulation of norepinephrine (NE)-producing neurons within the rodent locus coeruleus that co-express the endogenous opioid gene prodynorphin (Pdyn). These studies showcase ConVERGD as a versatile tool for targeting diverse cell types and reveal Pdyn-expressing NE+ locus coeruleus neurons as a small neuronal subpopulation capable of driving anxiogenic behavioral responses in rodents.
-
Histone Lysine Demethylase 4 Family Proteins Maintain the Transcriptional Program and Adrenergic Cellular State of MYCN-amplified NeuroblastomaCell Reports Medicine 2024
Neuroblastoma with MYCN amplification (MNA) is a high-risk disease that has a poor survival rate. Neuroblastoma displays cellular heterogeneity, including more differentiated (adrenergic) and more primitive (mesenchymal) cellular states. Here, we demonstrate that MYCN oncoprotein promotes a cellular state switch in mesenchymal cells to an adrenergic state, accompanied by induction of histone lysine demethylase 4 family members (KDM4A-C) that act in concert to control the expression of MYCN and adrenergic core regulatory circulatory (CRC) transcription factors. Pharmacologic inhibition of KDM4 blocks expression of MYCN and the adrenergic CRC transcriptome with genome-wide induction of transcriptionally repressive H3K9me3, resulting in potent anticancer activity against neuroblastomas with MNA by inducing neuroblastic differentiation and apoptosis. Furthermore, a short-term KDM4 inhibition in combination with conventional, cytotoxic chemotherapy results in complete tumor responses of xenografts with MNA. Thus, KDM4 blockade may serve as a transformative strategy to target the adrenergic CRC dependencies in MNA neuroblastomas.
-
Functional Characterization of Cooperating MGA Mutations in RUNX1::RUNX1T1 Acute Myeloid LeukemiaLeukemia 2024
MGA (Max-gene associated) is a dual-specificity transcription factor that negatively regulates MYC-target genes to inhibit proliferation and promote differentiation. Loss-of-function mutations in MGA have been commonly identified in several hematological neoplasms, including acute myeloid leukemia (AML) with RUNX1::RUNX1T1, however, very little is known about the impact of these MGA alterations on normal hematopoiesis or disease progression. We show that representative MGA mutations identified in patient samples abolish protein-protein interactions and transcriptional activity. Using a series of human and mouse model systems, including a newly developed conditional knock-out mouse strain, we demonstrate that loss of MGA results in upregulation of MYC and E2F targets, cell cycle genes, mTOR signaling, and oxidative phosphorylation in normal hematopoietic cells, leading to enhanced proliferation. The loss of MGA induces an open chromatin state at promoters of genes involved in cell cycle and proliferation. RUNX1::RUNX1T1 expression in Mga-deficient murine hematopoietic cells leads to a more aggressive AML with a significantly shortened latency. These data show that MGA regulates multiple pro-proliferative pathways in hematopoietic cells and cooperates with the RUNX1::RUNX1T1 fusion oncoprotein to enhance leukemogenesis.
-
RNA-binding Protein RBM5 Plays an Essential Role in Acute Myeloid Leukemia by Activating the Oncogenic Protein HOXA9Genome Biology 2024
The oncogenic protein HOXA9 plays a critical role in leukemia transformation and maintenance, and its aberrant expression is a hallmark of most aggressive acute leukemia. Although inhibiting the upstream regulators of HOXA9 has been proven as a significant therapeutic intervention, the comprehensive regulation network controlling HOXA9 expression in leukemia has not been systematically investigated.
-
THUMPD2 Catalyzes the N2-methylation of U6 snRNA of the Spliceosome Catalytic Center and Regulates Pre-mRNA Splicing and Retinal DegenerationNucleic Acids Res. 2024
The mechanisms by which the relatively conserved spliceosome manages the enormously large number of splicing events that occur in humans (∼200 000 versus ∼300 in yeast) are poorly understood. Here, we show deposition of one RNA modification-N2-methylguanosine (m2G) on the G72 of U6 snRNA (the catalytic center of the spliceosome) promotes efficient pre-mRNA splicing activity in human cells. This modification was identified to be conserved among vertebrates. Further, THUMPD2 was demonstrated as the methyltransferase responsible for U6 m2G72 by explicitly recognizing the U6-specific sequences and structural elements. The knock-out of THUMPD2 eliminated U6 m2G72 and impaired the pre-mRNA splicing activity, resulting in thousands of changed alternative splicing events of endogenous pre-mRNAs in human cells. Notably, the aberrantly spliced pre-mRNA population elicited the nonsense-mediated mRNA decay pathway. We further show that THUMPD2 was associated with age-related macular degeneration and retinal function. Our study thus demonstrates how an RNA epigenetic modification of the major spliceosome regulates global pre-mRNA splicing and impacts physiology and disease.
-
ABCC4 Impacts Megakaryopoiesis and Protects Megakaryocytes against 6-Mercaptopurine Induced CytotoxicityDrug Resist. Updates 2024
The role of ABCC4, an ATP-binding cassette transporter, in the process of platelet formation, megakaryopoiesis, is unknown. Here, we show that ABCC4 is highly expressed in megakaryocytes (MKs). Mining of public genomic data (ATAC-seq and genome wide chromatin interactions, Hi-C) revealed that key megakaryopoiesis transcription factors (TFs) interacted with ABCC4 regulatory elements and likely accounted for high ABCC4 expression in MKs. Importantly these genomic interactions for ABCC4 ranked higher than for genes with known roles in megakaryopoiesis suggesting a role for ABCC4 in megakaryopoiesis. We then demonstrate that ABCC4 is required for optimal platelet formation as in vitro differentiation of fetal liver derived MKs from Abcc4-/- mice exhibited impaired proplatelet formation and polyploidization, features required for optimal megakaryopoiesis. Likewise, a human megakaryoblastic cell line, MEG-01 showed that acute ABCC4 inhibition markedly suppressed key processes in megakaryopoiesis and that these effects were related to reduced cAMP export and enhanced dissociation of a negative regulator of megakaryopoiesis, protein kinase A (PKA) from ABCC4. PKA activity concomitantly increased after ABCC4 inhibition which was coupled with significantly reduced GATA-1 expression, a TF needed for optimal megakaryopoiesis. Further, ABCC4 protected MKs from 6-mercaptopurine (6-MP) as Abcc4-/- mice show a profound reduction in MKs after 6-MP treatment. In total, our studies show that ABCC4 not only protects the MKs but is also required for maximal platelet production from MKs, suggesting modulation of ABCC4 function might be a potential therapeutic strategy to regulate platelet production.
2023:12
-
PAX3-FOXO1 Dictates Myogenic Reprogramming and Rhabdomyosarcoma Identity in Endothelial ProgenitorsNat Commun 2023
Fusion-positive rhabdomyosarcoma (FP-RMS) driven by the expression of the PAX3-FOXO1 (P3F) fusion oncoprotein is an aggressive subtype of pediatric rhabdomyosarcoma. FP-RMS histologically resembles developing muscle yet occurs throughout the body in areas devoid of skeletal muscle highlighting that FP-RMS is not derived from an exclusively myogenic cell of origin. Here we demonstrate that P3F reprograms mouse and human endothelial progenitors to FP-RMS. We show that P3F expression in aP2-Cre expressing cells reprograms endothelial progenitors to functional myogenic stem cells capable of regenerating injured muscle fibers. Further, we describe a FP-RMS mouse model driven by P3F expression and Cdkn2a loss in endothelial cells. Additionally, we show that P3F expression in TP53-null human iPSCs blocks endothelial-directed differentiation and guides cells to become myogenic cells that form FP-RMS tumors in immunocompromised mice. Together these findings demonstrate that FP-RMS can originate from aberrant development of non-myogenic cells driven by P3F.
-
Acute Myeloid Leukemias with UBTF Tandem Duplications Are Sensitive to Menin InhibitorsBlood 2023
UBTF tandem duplications (UBTF-TDs) have recently emerged as a recurrent alteration in pediatric and adult acute myeloid leukemia (AML). UBTF-TD leukemias are characterized by a poor response to conventional chemotherapy and a transcriptional signature that mirrors NUP98-rearranged and NPM1-mutant AMLs, including HOX gene dysregulation. However, the mechanism of how UBTF-TD drives leukemogenesis remains unknown. In this study, we investigated the genomic occupancy of UBTF-TD in transformed cord-blood CD34+ (cbCD34+) cells and patient-derived xenograft models. We found that UBTF-TD protein maintained genomic occupancy at ribosomal DNA (rDNA) loci while also occupying genomic targets commonly dysregulated in UBTF-TD myeloid malignancies, such as the HOXA/HOXB gene clusters and MEIS1. These data suggest that UBTF-TD is a gain-of-function alteration that results in mislocalization to genomic loci dysregulated in UBTF-TD leukemias. UBTF-TD also co-occupies key genomic loci with KMT2A and Menin, which are known to be key partners involved in HOX-dysregulated leukemias. Using a protein degradation system, we showed that stemness, proliferation, and transcriptional signatures are dependent on sustained UBTF-TD localization to chromatin. Finally, we demonstrate that primary cells from UBTF-TD leukemias are sensitive to the Menin inhibitor SNDX-5613, resulting in markedly reduced in vitro and in vivo tumor growth, myeloid differentiation, and abrogation of the UBTF-TD leukemic expression signature. These findings provide a viable therapeutic strategy for patients with this high-risk AML subtype.
-
SNIP1 and PRC2 Coordinate Cell Fates of Neural Progenitors during Brain DevelopmentNat Commun 2023
Stem cell survival versus death is a developmentally programmed process essential for morphogenesis, sizing, and quality control of genome integrity and cell fates. Cell death is pervasive during development, but its programming is little known. Here, we report that Smad nuclear interacting protein 1 (SNIP1) promotes neural progenitor cell survival and neurogenesis and is, therefore, integral to brain development. The SNIP1-depleted brain exhibits dysplasia with robust induction of caspase 9-dependent apoptosis. Mechanistically, SNIP1 regulates target genes that promote cell survival and neurogenesis, and its activities are influenced by TGFβ and NFκB signaling pathways. Further, SNIP1 facilitates the genomic occupancy of Polycomb complex PRC2 and instructs H3K27me3 turnover at target genes. Depletion of PRC2 is sufficient to reduce apoptosis and brain dysplasia and to partially restore genetic programs in the SNIP1-depleted brain in vivo. These findings suggest a loci-specific regulation of PRC2 and H3K27 marks to toggle cell survival and death in the developing brain.
-
PP2Ac Deficiency Enhances Tumor Immunogenicity by Activating STING-Type I Interferon Signaling in GlioblastomaCancer Res 2023
Glioblastoma (GBM) is an immunologically “cold” tumor that does not respond to current immunotherapy. Here, we demonstrate a fundamental role for the α-isoform of the catalytic subunit of protein phosphatase-2A (PP2Ac) in regulating glioma immunogenicity. Genetic ablation of PP2Ac in glioma cells enhanced double stranded DNA (dsDNA) production and cGAS-type I interferon (IFN) signaling, MHC-I expression, and tumor mutational burden. In co-culture experiments, PP2Ac deficiency in glioma cells promoted dendritic cell (DC) cross presentation and clonal expansion of CD8+ T cells. In vivo, PP2Ac depletion sensitized tumors to immune checkpoint blockade and radiotherapy treatment. Single cell analysis demonstrated that PP2Ac deficiency increased CD8+ T cell, NK cell, and DC accumulation and reduced immunosuppressive tumor associated macrophages. Furthermore, loss of PP2Ac increased IFN signaling in myeloid and tumor cells and reduced expression of a tumor gene signature associated with worse patient survival in TCGA. Collectively, this study establishes a novel role for PP2Ac in inhibiting dsDNA-cGAS-STING signaling to suppress anti-tumor immunity in glioma.
-
Interrogating Bromodomain Inhibitor Resistance in KMT2A-rearranged Leukemia through Combinatorial CRISPR ScreensProceedings of the National Academy of Sciences 2023
Bromo- and extra-terminal domain inhibitors (BETi) have exhibited therapeutic activities in many cancers. However, the mechanisms controlling BETi response and resistance are not well understood. We conducted genome-wide loss-of-function CRISPR screens using BETi-treated KMT2A-rearranged (KMT2A-r) cell lines. We revealed that Speckle-type POZ protein (SPOP) gene (Speckle Type BTB/POZ Protein) deficiency caused significant BETi resistance, which was further validated in cell lines and xenograft models. Proteomics analysis and a kinase-vulnerability CRISPR screen indicated that cells treated with BETi are sensitive to GSK3 perturbation. Pharmaceutical inhibition of GSK3 reversed the BETi-resistance phenotype. Based on this observation, a combination therapy regimen inhibiting both BET and GSK3 was developed to impede KMT2A-r leukemia progression in patient-derived xenografts in vivo. Our results revealed molecular mechanisms underlying BETi resistance and a promising combination treatment regimen of ABBV-744 and CHIR-98014 by utilizing unique ex vivo and in vivo KMT2A-r PDX models.
-
How Estrogen, Testosterone, and Sex Differences Influence Serum Immunoglobulin Isotype Patterns in Mice and HumansViruses 2023
Females often exhibit superior immune responses compared to males toward vaccines and pathogens such as influenza viruses and SARS-CoV-2. To help explain these differences, we first studied serum immunoglobulin isotype patterns in C57BL/6 male and female mice. We focused on IgG2b, an isotype that lends to virus control and that has been previously shown to be elevated in murine females compared to males. Improvements in IgG2b serum levels, and/or IgG2b ratios with other non-IgM isotypes, were observed when: (i) wildtype (WT) female mice were compared to estrogen receptor knockout mice (IgG2b, IgG2b/IgG3, IgG2b/IgG1, and IgG2b/IgA were all higher in WT mice), (ii) unmanipulated female mice were compared to ovariectomized mice (IgG2b/IgA was higher in unmanipulated animals), (iii) female mice were supplemented with estrogen in the context of an inflammatory insult (IgG2b and IgG2b/IgG3 were improved by estrogen supplementation), and (iv) male mice were supplemented with testosterone, a hormone that can convert to estrogen in vivo (IgG2b, IgG2b/IgG3, IgG2b/IgG1, and IgG2b/IgA were all improved by supplementation). We next examined data from three sets of previously described male and female human blood samples. In each case, there were higher IgG2 levels, and/or ratios of IgG2 with non-IgM isotypes, in human females compared to males. The effects of sex and sex hormones in the mouse and human studies were subtle, but frequent, suggesting that sex hormones represent only a fraction of the factors that influence isotype patterns. Examination of the gene loci suggested that upregulation of murine IgG2b or human IgG2 could be mediated by estrogen receptor binding to estrogen response elements and cytosine-adenine (CA) repeats upstream of respective Cγ genes. Given that murine IgG2b and human IgG2 lend to virus control, the isotype biases in females may be sufficient to improve outcomes following vaccination or infection. Future attention to sex hormone levels, and consequent immunoglobulin isotype patterns, in clinical trials are encouraged to support the optimization of vaccine and drug products for male and female hosts.
-
Proteasome Inhibition Targets the KMT2A Transcriptional Complex in Acute Lymphoblastic LeukemiaNat Commun 2023
Rearrangments in Histone-lysine-N-methyltransferase 2A (KMT2Ar) are associated with pediatric, adult and therapy-induced acute leukemias. Infants with KMT2Ar acute lymphoblastic leukemia (ALL) have a poor prognosis with an event-free-survival of 38%. Herein we evaluate 1116 FDA approved compounds in primary KMT2Ar infant ALL specimens and identify a sensitivity to proteasome inhibition. Upon exposure to this class of agents, cells demonstrate a depletion of histone H2B monoubiquitination (H2Bub1) and histone H3 lysine 79 dimethylation (H3K79me2) at KMT2A target genes in addition to a downregulation of the KMT2A gene expression signature, providing evidence that it targets the KMT2A transcriptional complex and alters the epigenome. A cohort of relapsed/refractory KMT2Ar patients treated with this approach on a compassionate basis had an overall response rate of 90%. In conclusion, we report on a high throughput drug screen in primary pediatric leukemia specimens whose results translate into clinically meaningful responses. This innovative treatment approach is now being evaluated in a multi-institutional upfront trial for infants with newly diagnosed ALL.
-
PP2Ac/STRN4 Negatively Regulates STING-Type I Interferon Signaling in Tumor Associated MacrophagesJ Clin Invest 2023
Stimulator of IFN genes type I (STING-Type I) IFN signaling in myeloid cells plays a critical role in effective antitumor immune responses, but STING agonists as monotherapy have shown limited efficacy in clinical trials. The mechanisms that downregulate STING signaling are not fully understood. Here, we report that protein phosphatase 2A (PP2A), with its specific B regulatory subunit Striatin 4 (STRN4), negatively regulated STING-Type I IFN in macrophages. Mice with macrophage PP2A deficiency exhibited reduced tumor progression. The tumor microenvironment showed decreased immunosuppressive and increased IFN-activated macrophages and CD8+ T cells. Mechanistically, we demonstrated that Hippo kinase MST1/2 was required for STING activation. STING agonists induced dissociation of PP2A from MST1/2 in normal macrophages, but not in tumor conditioned macrophages. Furthermore, our data showed that STRN4 mediated PP2A binding to and dephosphorylation of Hippo kinase MST1/2, resulting in stabilization of YAP/TAZ to antagonize STING activation. In human patients with glioblastoma (GBM), YAP/TAZ was highly expressed in tumor-associated macrophages but not in nontumor macrophages. We also demonstrated that PP2A/STRN4 deficiency in macrophages reduced YAP/TAZ expression and sensitized tumor-conditioned macrophages to STING stimulation. In summary, we demonstrated that PP2A/STRN4-YAP/TAZ has, in our opinion, been an unappreciated mechanism that mediates immunosuppression in tumor-associated macrophages, and targeting the PP2A/STRN4-YAP/TAZ axis can sensitize tumors to immunotherapy.
-
Spatiotemporal Regulation of Cholangiocarcinoma Growth and Dissemination by Peritumoral Myofibroblasts in a Vcam1-dependent MannerOncogene 2023
Intrahepatic cholangiocarcinoma (iCCA) is characterized by its highly desmoplastic stroma. Myofibroblasts (MFs) are present both within the tumor mass (intratumoral MFs, iMFs) and at the tumor border (peritumoral MFs, pMFs). Using a spheroid-based coculture system, we show that the initial iCCA-pMF contact is growth suppressive to the tumor cells. However, prolonged iCCA-pMF interaction elicits significant tumor cell invasion and dissemination. We find that vascular cell adhesion molecule-1 (Vcam1) level is elevated in tumor cells in contact with pMFs but low in disseminated tumor cells both in vitro and in vivo. A gene regulatory network analysis of mouse and patient iCCA tumors and Vcam1 knockout (Vcam1KO) demonstrate a heavy involvement of Vcam1 in epithelial-to-mesenchymal transition. While Vcam1KO has only a limited impact on tumor cell growth in their monoculture, Vcam1KO spheroids exhibit instant dissemination and a severe growth defect when cocultured with pMFs. When transplanted into the liver, Vcam1KO iCCA cells show a similar increase in dissemination but a significant defect in establishing primary and metastatic tumors. Incomplete blocking of Vcam1 in vivo reduces the size but increase the number of metastatic lesions. Overall, our study shows a spatiotemporal regulation of iCCA growth and dissemination by pMFs in a Vcam1-dependent manner.
-
Auxin-Inducible Degron 2 System Deciphers Functions of CTCF Domains in Transcriptional RegulationGenome Biol. 2023
CTCF is a well-established chromatin architectural protein that also plays various roles in transcriptional regulation. While CTCF biology has been extensively studied, how the domains of CTCF function to regulate transcription remains unknown. Additionally, the original auxin-inducible degron 1 (AID1) system has limitations in investigating the function of CTCF.
-
Modulation of Protease Expression by the Transcription Factor Ptx1/PITX Regulates Protein Quality Control during AgingCell Reports 2023
Protein quality control is important for healthy aging and is dysregulated in age-related diseases. The autophagy-lysosome and ubiquitin-proteasome are key for proteostasis, but it remains largely unknown whether other proteolytic systems also contribute to maintain proteostasis during aging. Here, we find that expression of proteolytic enzymes (proteases/peptidases) distinct from the autophagy-lysosome and ubiquitin-proteasome systems declines during skeletal muscle aging in Drosophila. Age-dependent protease downregulation undermines proteostasis, as demonstrated by the increase in detergent-insoluble poly-ubiquitinated proteins and pathogenic huntingtin-polyQ levels in response to protease knockdown. Computational analyses identify the transcription factor Ptx1 (homologous to human PITX1/2/3) as a regulator of protease expression. Consistent with this model, Ptx1 protein levels increase with aging, and Ptx1 RNAi counteracts the age-associated downregulation of protease expression. Moreover, Ptx1 RNAi improves muscle protein quality control in a protease-dependent manner and extends lifespan. These findings indicate that proteases and their transcriptional modulator Ptx1 ensure proteostasis during aging.
-
ZBTB18 Restricts Chromatin Accessibility and Prevents Transcriptional Adaptations That Drive MetastasisScience Advances 2023
Metastases arise from rare cancer cells that successfully adapt to the diverse microenvironments encountered during dissemination through the bloodstream and colonization of distant tissues. How cancer cells acquire the ability to appropriately respond to microenvironmental stimuli remains largely unexplored. Here, we report an epigenetic pliancy mechanism that allows cancer cells to successfully metastasize. We find that a decline in the activity of the transcriptional repressor ZBTB18 defines metastasis-competent cancer cells in mouse models. Restoration of ZBTB18 activity reduces chromatin accessibility at the promoters of genes that drive metastasis, such as Tgfbr2, and this prevents TGFβ1 pathway activation and consequently reduces cell migration and invasion. Besides repressing the expression of metastatic genes, ZBTB18 also induces widespread chromatin closing, a global epigenetic adaptation previously linked to reduced phenotypic flexibility. Thus, ZBTB18 is a potent chromatin regulator, and the loss of its activity enhances chromatin accessibility and transcriptional adaptations that promote the phenotypic changes required for metastasis.
2022:8
-
Epigenetic Activation of the FLT3 Gene by ZNF384 Fusion Confers a Therapeutic Susceptibility in Acute Lymphoblastic LeukemiaNat Commun 2022
FLT3 is an attractive therapeutic target in acute lymphoblastic leukemia (ALL) but the mechanism for its activation in this cancer is incompletely understood. Profiling global gene expression in large ALL cohorts, we identify over-expression of FLT3 in ZNF384-rearranged ALL, consistently across cases harboring different fusion partners with ZNF384. Mechanistically, we discover an intergenic enhancer element at the FLT3 locus that is exclusively activated in ZNF384-rearranged ALL, with the enhancer-promoter looping directly mediated by the fusion protein. There is also a global enrichment of active enhancers within ZNF384 binding sites across the genome in ZNF384-rearranged ALL cells. Downregulation of ZNF384 blunts FLT3 activation and decreases ALL cell sensitivity to FLT3 inhibitor gilteritinib in vitro. In patient-derived xenograft models of ZNF384-rearranged ALL, gilteritinib exhibits significant anti-leukemia efficacy as a monotherapy in vivo. Collectively, our results provide insights into FLT3 regulation in ALL and point to potential genomics-guided targeted therapy for this patient population.
-
Targeting KDM4 for Treating PAX3-FOXO1–Driven Alveolar RhabdomyosarcomaScience Translational Medicine 2022
Chimeric transcription factors drive lineage-specific oncogenesis but are notoriously difficult to target. Alveolar rhabdomyosarcoma (RMS) is an aggressive childhood soft tissue sarcoma transformed by the pathognomonic Paired Box 3–Forkhead Box O1 (PAX3-FOXO1) fusion protein, which governs a core regulatory circuitry transcription factor network. Here, we show that the histone lysine demethylase 4B (KDM4B) is a therapeutic vulnerability for PAX3-FOXO1+ RMS. Genetic and pharmacologic inhibition of KDM4B substantially delayed tumor growth. Suppression of KDM4 proteins inhibited the expression of core oncogenic transcription factors and caused epigenetic alterations of PAX3-FOXO1–governed superenhancers. Combining KDM4 inhibition with cytotoxic chemotherapy led to tumor regression in preclinical PAX3-FOXO1+ RMS subcutaneous xenograft models. In summary, we identified a targetable mechanism required for maintenance of the PAX3-FOXO1–related transcription factor network, which may translate to a therapeutic approach for fusion-positive RMS.
-
ZNF384 Fusion Oncoproteins Drive Lineage Aberrancy in Acute LeukemiaBlood Cancer Discovery 2022
ZNF384-rearranged fusion oncoproteins (FO) define a subset of lineage ambiguous leukemias, but their mechanistic role in leukemogenesis and lineage ambiguity is poorly understood. Using viral expression in mouse and human hematopoietic stem and progenitor cells (HSPC) and a Ep300::Znf384 knockin mouse model, we show that ZNF384 FO promote hematopoietic expansion, myeloid lineage skewing, and self-renewal. In mouse HSPCs, concomitant lesions, such as NRASG12D, were required for fully penetrant leukemia, whereas in human HSPCs, expression of ZNF384 FO drove B/myeloid leukemia, with sensitivity of a ZNF384-rearranged xenograft to FLT3 inhibition in vivo. Mechanistically, ZNF384 FO occupy a subset of predominantly intragenic/enhancer regions with increased histone 3 lysine acetylation and deregulate expression of hematopoietic stem cell transcription factors. These data define a paradigm for FO-driven lineage ambiguous leukemia, in which expression in HSPCs results in deregulation of lineage-specific genes and hematopoietic skewing, progressing to full leukemia in the context of proliferative stress.Expression of ZNF384 FO early in hematopoiesis results in binding and deregulation of key hematopoietic regulators, skewing of hematopoiesis, and priming for leukemic transformation. These results reveal the interplay between cell of origin and expression of ZNF384 FO to mediate lineage ambiguity and leukemia development.This article is highlighted in the In This Issue feature, p. 171
-
Molecular Mechanisms of ARID5B-Mediated Genetic Susceptibility to Acute Lymphoblastic LeukemiaJNCI: Journal of the National Cancer Institute 2022
There is growing evidence for the inherited basis of susceptibility to childhood acute lymphoblastic leukemia (ALL). Genome-wide association studies have identified non-coding ALL risk variants at the ARID5B gene locus, but their exact functional effects and the molecular mechanism linking ARID5B to B-cell ALL leukemogenesis remain largely unknown.We performed targeted sequencing of ARID5B in germline DNA of 5008 children with ALL. Variants were evaluated for association with ALL susceptibility using 3644 patients from the UK10K cohort as non-ALL controls, under an additive model. Cis-regulatory elements in ARID5B were systematically identified using dCas9-KRAB–mediated enhancer interference system enhancer screen in ALL cells. Disruption of transcription factor binding by ARID5B variant was predicted informatically and then confirmed using chromatin immunoprecipitation and coimmunoprecipitation. ARID5B variant association with hematological traits was examined using UK Biobank dataset. All statistical tests were 2-sided.We identified 54 common variants in ARID5B statistically significantly associated with leukemia risk, all of which were noncoding. Six cis-regulatory elements at the ARID5B locus were discovered using CRISPR-based high-throughput enhancer screening. Strikingly, the top ALL risk variant (rs7090445, P\,= 5.57\,\texttimes 10–45) is located precisely within the strongest enhancer element, which is also distally tethered to the ARID5B promoter. The variant allele disrupts the MEF2C binding motif sequence, resulting in reduced MEF2C affinity and decreased local chromosome accessibility. MEF2C influences ARID5B expression in ALL, likely via a transcription factor complex with RUNX1. Using the UK Biobank dataset (n\,= 349\mkern1mu861), we showed that rs7090445 was also associated with lymphocyte percentage and count in the general population (P\,= 8.6\,\texttimes 10–22 and 2.1\,\texttimes 10–18, respectively).Our results indicate that ALL risk variants in ARID5B function by modulating cis-regulatory elements at this locus.
-
NSD1 Mediates Antagonism between SWI/SNF and Polycomb Complexes and Is Required for Transcriptional Activation upon EZH2 InhibitionMolecular Cell 2022
Disruption of antagonism between SWI/SNF chromatin remodelers and polycomb repressor complexes drives the formation of numerous cancer types. Recently, an inhibitor of the polycomb protein EZH2 was approved for the treatment of a sarcoma mutant in the SWI/SNF subunit SMARCB1, but resistance occurs. Here, we performed CRISPR screens in SMARCB1-mutant rhabdoid tumor cells to identify genetic contributors to SWI/SNF-polycomb antagonism and potential resistance mechanisms. We found that loss of the H3K36 methyltransferase NSD1 caused resistance to EZH2 inhibition. We show that NSD1 antagonizes polycomb via cooperation with SWI/SNF and identify co-occurrence of NSD1 inactivation in SWI/SNF-defective cancers, indicating in vivo relevance. We demonstrate that H3K36me2 itself has an essential role in the activation of polycomb target genes as inhibition of the H3K36me2 demethylase KDM2A restores the efficacy of EZH2 inhibition in SWI/SNF-deficient cells lacking NSD1. Together our data expand the mechanistic understanding of SWI/SNF and polycomb interplay and identify NSD1 as the key for coordinating this transcriptional control.
-
The Myokine Fibcd1 Is an Endogenous Determinant of Myofiber Size and Mitigates Cancer-Induced Myofiber AtrophyNat Commun 2022
Decline in skeletal muscle cell size (myofiber atrophy) is a key feature of cancer-induced wasting (cachexia). In particular, atrophy of the diaphragm, the major muscle responsible for breathing, is an important determinant of cancer-associated mortality. However, therapeutic options are limited. Here, we have used Drosophila transgenic screening to identify muscle-secreted factors (myokines) that act as paracrine regulators of myofiber growth. Subsequent testing in mouse myotubes revealed that mouse Fibcd1 is an evolutionary-conserved myokine that preserves myofiber size via ERK signaling. Local administration of recombinant Fibcd1 (rFibcd1) ameliorates cachexia-induced myofiber atrophy in the diaphragm of mice bearing patient-derived melanoma xenografts and LLC carcinomas. Moreover, rFibcd1 impedes cachexia-associated transcriptional changes in the diaphragm. Fibcd1-induced signaling appears to be muscle selective because rFibcd1 increases ERK activity in myotubes but not in several cancer cell lines tested. We propose that rFibcd1 may help reinstate myofiber size in the diaphragm of patients with cancer cachexia.
-
Genomic Profiling Identifies Genes and Pathways Dysregulated by HEY1-NCOA2 Fusion and Shines a Light on Mesenchymal Chondrosarcoma TumorigenesisJ Pathol 2022
Mesenchymal chondrosarcoma is a rare, high-grade, primitive mesenchymal tumor. It accounts for around 2-10% of all chondrosarcomas and mainly affects adolescents and young adults. We previously described the HEY1-NCOA2 as a recurrent gene fusion in mesenchymal chondrosarcoma, an important breakthrough for characterizing this disease; however, little study had been done to characterize the fusion protein functionally, in large part due to a lack of suitable models for evaluating the impact of HEY1-NCOA2 expression in the appropriate cellular context. We used iPSC-derived mesenchymal stem cells (iPSC-MSCs), which can differentiate into chondrocytes, and generated stable transduced iPSC-MSCs with inducible expression of HEY1-NCOA2 fusion protein, wildtype HEY1 or wildtype NCOA2. We next comprehensively analyzed both the DNA binding properties and transcriptional impact of HEY1-NCOA2 expression by integrating genome-wide chromatin immunoprecipitation sequencing (ChIP-seq) and expression profiling (RNA-seq). We demonstrated that HEY1-NCOA2 fusion protein preferentially binds to promoter regions of canonical HEY1 targets, resulting in transactivation of HEY1 targets, and significantly enhances cell proliferation. Intriguingly, we identified that both PDGFB and PDGFRA were directly targeted and upregulated by HEY1-NCOA2; and the fusion protein, but not wildtype HEY1 or NCOA2, dramatically increased the level of phospho-AKT (Ser473). Our findings provide a rationale for exploring PDGF/PI3K/AKT inhibition in treating mesenchymal chondrosarcoma. \textcopyright 2022 The Authors. The Journal of Pathology published by John Wiley & Sons Ltd on behalf of The Pathological Society of Great Britain and Ireland.
-
PROSER1 Mediates TET2 O-GlcNAcylation to Regulate DNA Demethylation on UTX-Dependent Enhancers and CpG IslandsLife Science Alliance 2022
DNA methylation at enhancers and CpG islands usually leads to gene repression, which is counteracted by DNA demethylation through the TET protein family. However, how TET enzymes are recruited and regulated at these genomic loci is not fully understood. Here, we identify TET2, the glycosyltransferase OGT and a previously undescribed proline and serine rich protein, PROSER1 as interactors of UTX, a component of the enhancer-associated MLL3/4 complexes. We find that PROSER1 mediates the interaction between OGT and TET2, thus promoting TET2 O-GlcNAcylation and protein stability. In addition, PROSER1, UTX, TET1/2, and OGT colocalize on many genomic elements genome-wide. Loss of PROSER1 results in lower enrichment of UTX, TET1/2, and OGT at enhancers and CpG islands, with a concomitant increase in DNA methylation and transcriptional down-regulation of associated target genes and increased DNA hypermethylation encroachment at H3K4me1-predisposed CpG islands. Furthermore, we provide evidence that PROSER1 acts as a more general regulator of OGT activity by controlling O-GlcNAcylation of multiple other chromatin signaling pathways. Taken together, this study describes for the first time a regulator of TET2 O-GlcNAcylation and its implications in mediating DNA demethylation at UTX-dependent enhancers and CpG islands and supports an important role for PROSER1 in regulating the function of various chromatin-associated proteins via OGT-mediated O-GlcNAcylation.
2021:9
-
Control of Foxp3 Induction and Maintenance by Sequential Histone Acetylation and DNA DemethylationCell Report 2021
Regulatory T (Treg) cells play crucial roles in suppressing deleterious immune response. Here, we investigate how Treg cells are mechanistically induced in vitro (iTreg) and stabilized via transcriptional regulation of Treg lineage-specifying factor Foxp3. We find that acetylation of histone tails at the Foxp3 promoter is required for inducing Foxp3 transcription. Upon induction, histone acetylation signals via bromodomain-containing proteins, particularly targets of inhibitor JQ1, and sustains Foxp3 transcription via a global or trans effect. Subsequently, Tet-mediated DNA demethylation of Foxp3 cis-regulatory elements, mainly enhancer CNS2, increases chromatin accessibility and protein binding, stabilizing Foxp3 transcription and obviating the need for the histone acetylation signal. These processes transform stochastic iTreg induction into a stable cell fate, with the former sensitive and the latter resistant to genetic and environmental perturbations. Thus, sequential histone acetylation and DNA demethylation in Foxp3 induction and maintenance reflect stepwise mechanical switches governing iTreg cell lineage specification.
-
A Dual Role of Human tRNA Methyltransferase hTrmt13 in Regulating Translation and TranscriptionEMBO J 2021
Since numerous RNAs and RBPs prevalently localize to active chromatin regions, many RNA-binding proteins (RBPs) may be potential transcriptional regulators. RBPs are generally thought to regulate transcription via noncoding RNAs. Here, we describe a distinct, dual mechanism of transcriptional regulation by the previously uncharacterized tRNA-modifying enzyme, hTrmt13. On one hand, hTrmt13 acts in the cytoplasm to catalyze 2’-O-methylation of tRNAs, thus regulating translation in a manner depending on its tRNA-modification activity. On the other hand, nucleus-localized hTrmt13 directly binds DNA as a transcriptional co-activator of key epithelial-mesenchymal transition factors, thereby promoting cell migration independent of tRNA-modification activity. These dual functions of hTrmt13 are mutually exclusive, as it can bind either DNA or tRNA through its CHHC zinc finger domain. Finally, we find that hTrmt13 expression is tightly associated with poor prognosis and survival in diverse cancer patients. Our discovery of the noncatalytic roles of an RNA-modifying enzyme provides a new perspective for understanding epitranscriptomic regulation.
-
KDM6B Promotes Activation of the Oncogenic CDK4/6-pRB-E2F Pathway by Maintaining Enhancer Activity in MYCN-amplified NeuroblastomaNature Communication 2021
The H3K27me2/me3 histone demethylase KDM6B is essential to neuroblastoma cell survival. However, the mechanism of KDM6B action remains poorly defined. We demonstrate that inhibition of KDM6B activity 1) reduces the chromatin accessibility of E2F target genes and MYCN, 2) selectively leads to an increase of H3K27me3 but a decrease of the enhancer mark H3K4me1 at the CTCF and BORIS binding sites, which may, consequently, disrupt the long-range chromatin interaction of MYCN and E2F target genes, and 3) phenocopies the transcriptome induced by the specific CDK4/6 inhibitor palbociclib. Overexpression of CDK4/6 or Rb1 knockout confers neuroblastoma cell resistance to both palbociclib and the KDM6 inhibitor GSK-J4. These data indicate that KDM6B promotes an oncogenic CDK4/6-pRB-E2F pathway in neuroblastoma cells via H3K27me3-dependent enhancer-promoter interactions, providing a rationale to target KDM6B for high-risk neuroblastoma.
-
Acute Depletion of CTCF Rewires Genome-Wide Chromatin AccessibilityGenome Biology 2021
Background The transcription factor CTCF appears indispensable in defining topologically associated domain boundaries and maintaining chromatin loop structures within these domains, supported by numerous functional studies. However, acute depletion of CTCF globally reduces chromatin interactions but does not significantly alter transcription. Results Here, we systematically integrate multi-omics data including ATAC-seq, RNA-seq, WGBS, Hi-C, Cut&Run, and CRISPR-Cas9 survival dropout screens, and time-solved deep proteomic and phosphoproteomic analyses in cells carrying auxin-induced degron at endogenous CTCF locus. Acute CTCF protein degradation markedly rewires genome-wide chromatin accessibility. Increased accessible chromatin regions are frequently located adjacent to CTCF-binding sites at promoter regions and insulator sites associated with enhanced transcription of nearby genes. In addition, we use CTCF-associated multi-omics data to establish a combinatorial data analysis pipeline to discover CTCF co-regulatory partners. We successfully identify 40 candidates, including multiple established partners. Interestingly, many CTCF co-regulators that have alterations of their respective downstream gene expression do not show changes of their own expression levels across the multi-omics measurements upon acute CTCF loss, highlighting the strength of our system to discover hidden co-regulatory partners associated with CTCF-mediated transcription. Conclusions This study highlights that CTCF loss rewires genome-wide chromatin accessibility, which plays a critical role in transcriptional regulation.
-
Enhancer Hijacking Drives Oncogenic BCL11B Expression in Lineage Ambiguous Stem Cell LeukemiaCancer Discovery 2021
Lineage ambiguous leukemias are high-risk malignancies of poorly understood genetic basis. Here, we describe a distinct subgroup of acute leukemia with expression of myeloid, T lymphoid and stem cell markers driven by aberrant allele-specific deregulation of BCL11B, a master transcription factor responsible for thymic T-lineage commitment and specification. Mechanistically, this deregulation was driven by chromosomal rearrangements that juxtapose BCL11B to super-enhancers active in hematopoietic progenitors, or focal amplifications that generate a super-enhancer from a non-coding element distal to BCL11B. Chromatin conformation analyses demonstrate long range interactions of rearranged enhancers with the expressed BCL11B allele, and association of BCL11B with activated hematopoietic progenitor cell cis-regulatory elements, suggesting BCL11B is aberrantly co-opted into a gene regulatory network that drives transformation by maintaining a progenitor state. These data support a role for ectopic BCL11B expression in primitive hematopoietic cells mediated by enhancer hijacking as an oncogenic driver of human lineage ambiguous leukemia.
-
Foxp3 Enhancers Synergize to Maximize Regulatory T Cell Suppressive CapacityJournal of Experimental Medicine 2021
T reg cells bearing a diverse antigen receptor repertoire suppress pathogenic T cells and maintain immune homeostasis during their long lifespan. How their robust function is determined genetically remains elusive. Here, we investigate the regulatory space of the cis-regulatory elements of T reg lineage–specifying factor Foxp3. Foxp3 enhancers are known as distinct readers of environmental cues controlling T reg cell induction or lineage stability. However, their single deficiencies cause mild, if any, immune dysregulation, leaving the key transcriptional mechanisms determining Foxp3 expression and thereby T reg cell suppressive capacity uncertain. We examined the collective activities of Foxp3 enhancers and found that they coordinate to maximize T reg cell induction, Foxp3 expression level, or lineage stability through distinct modes and that ablation of synergistic enhancers leads to lethal autoimmunity in young mice. Thus, the induction and maintenance of a diverse, stable T reg cell repertoire rely on combinatorial Foxp3 enhancers, suggesting broad, stage-specific, synergistic activities of cell-intrinsic factors and cell-extrinsic cues in determining T reg cell suppressive capacity.
-
A Distal Foxp3 Enhancer Enables Interleukin-2 Dependent Thymic Treg Cell Lineage Commitment for Robust Immune ToleranceImmunity 2021
Activation of the STAT5 transcription factor downstream of the Interleukin-2 receptor (IL-2R) induces expression of Foxp3, a critical step in the differentiation of regulatory T (Treg) cells. Due to the pleiotropic effects of IL-2R signaling, it is unclear how STAT5 acts directly on the Foxp3 locus to promote its expression. Here, we report that IL-2 – STAT5 signaling converged on an enhancer (CNS0) during Foxp3 induction. CNS0 facilitated the IL-2 dependent CD25+Foxp3– precursor to Treg cell transition in the thymus. Its deficiency resulted in impaired Treg cell generation in neonates, which was partially mitigated with age. While the thymic Treg cell paucity caused by CNS0 deficiency did not result in autoimmunity on its own, it exacerbated autoimmune manifestations caused by disruption of the Aire gene. Thus, CNS0 enhancer activity ensures robust Treg cell differentiation early in postnatal life and cooperatively with other tolerance mechanisms minimizes autoimmunity.
-
Proteasome Stress in Skeletal Muscle Mounts a Long-Range Protective Response That Delays Retinal and Brain AgingCell Metabolism 2021
Neurodegeneration in the central nervous system (CNS) is a defining feature of organismal aging that is influenced by peripheral tissues. Clinical observations indicate that skeletal muscle influences CNS aging, but the underlying muscle-to-brain signaling remains unexplored. In Drosophila, we find that moderate perturbation of the proteasome in skeletal muscle induces compensatory preservation of CNS proteostasis during aging. Such long-range stress signaling depends on muscle-secreted Amyrel amylase. Mimicking stress-induced Amyrel upregulation in muscle reduces age-related accumulation of poly-ubiquitinated proteins in the brain and retina via chaperones. Preservation of proteostasis stems from the disaccharide maltose, which is produced via Amyrel amylase activity. Correspondingly, RNAi for SLC45 maltose transporters reduces expression of Amyrel-induced chaperones and worsens brain proteostasis during aging. Moreover, maltose preserves proteostasis and neuronal activity in human brain organoids challenged by thermal stress. Thus, proteasome stress in skeletal muscle hinders retinal and brain aging by mounting an adaptive response via amylase/maltose.
-
MethylationToActivity: A Deep-Learning Framework That Reveals Promoter Activity Landscapes from DNA Methylomes in Individual TumorsGenome Biology 2021
Although genome-wide DNA methylomes have demonstrated their clinical value as reliable biomarkers for tumor detection, subtyping, and classification, their direct biological impacts at the individual gene level remain elusive. Here we present MethylationToActivity (M2A), a machine learning framework that uses convolutional neural networks to infer promoter activities based on H3K4me3 and H3K27ac enrichment, from DNA methylation patterns for individual genes. Using publicly available datasets in real-world test scenarios, we demonstrate that M2A is highly accurate and robust in revealing promoter activity landscapes in various pediatric and adult cancers, including both solid and hematologic malignant neoplasms.
2020:8
-
FBXO11-Mediated Proteolysis of BAHD1 Relieves PRC2-Dependent Transcriptional Repression in ErythropoiesisBlood 2020
The histone mark H3K27me3 and its reader/writer Polycomb repressive complex 2 (PRC2) mediate widespread transcriptional repression in stem and progenitor cells. Mechanisms that regulate this activity are critical for hematopoietic development but poorly understood. Here we show that the E3 ubiquitin ligase FBXO11 relieves PRC2-mediated repression during erythroid maturation by targeting its newly identified substrate BAHD1, an H3K27me3 reader that recruits transcriptional co-repressors. Erythroblasts lacking FBXO11 are developmentally delayed, with reduced expression of maturation-associated genes, most of which harbor bivalent histone marks (activating H3K4me3 and repressive H3K27me3), bind BAHD1, and fail to recruit the erythroid transcription factor GATA1. The BAHD1 complex interacts physically with PRC2 and depletion of either component restores FBXO11-deficient erythroid gene expression. Our studies identify BAHD1 as a novel effector of PRC2-mediated repression and reveal how a single E3 ubiquitin ligase eliminates PRC2 repression at developmentally poised bivalent genes during erythropoiesis.
-
Functional Interrogation of HOXA9 Regulome in MLLr Leukemia via Reporter-Based CRISPR/Cas9 ScreeneLife 2020
Aberrant HOXA9 expression is a hallmark of most aggressive acute leukemias, notably those with KMT2A (MLL) gene rearrangements. HOXA9 overexpression not only predicts poor diagnosis and outcome but also plays a critical role in leukemia transformation and maintenance. However, our current understanding of HOXA9 regulation in leukemia is limited, hindering development of therapeutic strategies. Here, we generated the HOXA9-mCherry knock-in reporter cell lines to dissect HOXA9 regulation. By utilizing the reporter and CRISPR/Cas9 screens, we identified transcription factors controlling HOXA9 expression, including a novel regulator, USF2, whose depletion significantly down-regulated HOXA9 expression and impaired MLLr leukemia cell proliferation. Ectopic expression of Hoxa9 rescued impaired leukemia cell proliferation upon USF2 loss. Cut and Run analysis revealed the direct occupancy of USF2 at HOXA9 promoter in MLLr leukemia cells. Collectively, the HOXA9 reporter facilitated the functional interrogation of the HOXA9 regulome and has advanced our understanding of the molecular regulation network in HOXA9-driven leukemia.
-
UTX/KDM6A Suppresses AP-1 and a Gliogenesis Program during Neural Differentiation of Human Pluripotent Stem CellsEpigenetics & Chromatin 2020
Background UTX/KDM6A is known to interact and influence multiple different chromatin modifiers to promote an open chromatin environment to facilitate gene activation, but its molecular activities in developmental gene regulation remain unclear. Results We report that in human neural stem cells, UTX binding correlates with both promotion and suppression of gene expression. These activities enable UTX to modulate neural stem cell self-renewal, promote neurogenesis, and suppress gliogenesis. In neural stem cells, UTX has a less influence over histone H3 lysine 27 and lysine 4 methylation but more predominantly affects histone H3 lysine 27 acetylation and chromatin accessibility. Furthermore, UTX suppresses components of AP-1 and, in turn, a gliogenesis program. Conclusions Our findings revealed that UTX coordinates dualistic gene regulation to govern neural stem cell properties and neurogenesis–gliogenesis switch.
-
Ybx1 Fine-Tunes PRC2 Activities to Control Embryonic Brain DevelopmentNature Communications 2020
Chromatin modifiers affect spatiotemporal gene expression programs that underlie organismal development. The Polycomb repressive complex 2 (PRC2) is a crucial chromatin modifier in executing neurodevelopmental programs. Here, we find that PRC2 interacts with the nucleic acid–binding protein Ybx1. In the mouse embryo in vivo, Ybx1 is required for forebrain specification and restricting mid-hindbrain growth. In neural progenitor cells (NPCs), Ybx1 controls self-renewal and neuronal differentiation. Mechanistically, Ybx1 highly overlaps PRC2 binding genome-wide, controls PRC2 distribution, and inhibits H3K27me3 levels. These functions are consistent with Ybx1-mediated promotion of genes involved in forebrain specification, cell proliferation, or neuronal differentiation. In Ybx1-knockout NPCs, H3K27me3 reduction by PRC2 enzymatic inhibitor or genetic depletion partially rescues gene expression and NPC functions. Our findings suggest that Ybx1 fine-tunes PRC2 activities to regulate spatiotemporal gene expression in embryonic neural development and uncover a crucial epigenetic mechanism balancing forebrain–hindbrain lineages and self-renewal–differentiation choices in NPCs.
-
From Influenza Virus Infections to Lupus: Synchronous Estrogen Receptor α and RNA Polymerase II Binding Within the Immunoglobulin Heavy Chain LocusViral Immunology 2020
Males and females respond to pathogens differently and exhibit significantly different frequencies of autoimmune disease. For example, vaccinated adult females control influenza virus better than males, but females suffer systemic lupus erythematosus at a 9:1 frequency compared to males. Numerous explanations have been offered for these sex differences, but most have involved indirect mechanisms by which estrogen, a nuclear hormone, modifies cell barriers or immunity. In search of a direct mechanism, we examined the binding of estrogen receptor α (ERα), a class I nuclear hormone receptor, to the immunoglobulin heavy chain locus. Here, we show that in purified murine B cells, ERα and RNA polymerase II (RNA Pol II) exhibit extraordinarily similar DNA binding patterns. We further demonstrate that ERα preferentially binds adenosine–cytidine (AC)-repeats in the immunoglobulin heavy chain locus when supplemental estrogen is added to purified, lipopolysaccharide-activated B cells. Based on these and previous data, we hypothesize that (i) estrogen guides the binding of ERα and its RNA Pol II partner within the locus, which in turn instructs sterile transcription and class switch recombination (CSR), (ii) ERα binding to AC-repeats modifies the DNA architecture and loops associated with CSR, and (iii) by these mechanisms, estrogen instructs antibody expression. By targeting ERα-DNA interactions in the immunoglobulin heavy chain locus, clinicians may ultimately enhance antibody responses in the context of infectious diseases and reduce antibody responses in the context of allergic or autoimmune reactions.
-
MYCN Amplification and ATRX Mutations Are Incompatible in NeuroblastomaNature Communications 2020
Aggressive cancers often have activating mutations in growth-controlling oncogenes and inactivating mutations in tumor-suppressor genes. In neuroblastoma, amplification of the MYCN oncogene and inactivation of the ATRX tumor-suppressor gene correlate with high-risk disease and poor prognosis. Here we show that ATRX mutations and MYCN amplification are mutually exclusive across all ages and stages in neuroblastoma. Using human cell lines and mouse models, we found that elevated MYCN expression and ATRX mutations are incompatible. Elevated MYCN levels promote metabolic reprogramming, mitochondrial dysfunction, reactive-oxygen species generation, and DNA-replicative stress. The combination of replicative stress caused by defects in the ATRX–histone chaperone complex, and that induced by MYCN-mediated metabolic reprogramming, leads to synthetic lethality. Therefore, ATRX and MYCN represent an unusual example, where inactivation of a tumor-suppressor gene and activation of an oncogene are incompatible. This synthetic lethality may eventually be exploited to improve outcomes for patients with high-risk neuroblastoma.
-
An ABC Transporter Drives Medulloblastoma Pathogenesis by Regulating Sonic Hedgehog SignalingCancer Research 2020
Mutations in Sonic hedgehog (SHH) signaling promote aberrant proliferation and tumor growth. SHH medulloblastoma (MB) is among the most frequent brain tumors in children less than three years of age. While key components of the SHH pathway are well-known, we hypothesized that new disease-modifying targets of SHH MB might be identified from large-scale bioinformatics and systems biology analyses. Using a data-driven systems biology approach, we built a medulloblastoma-specific interactome. The ATP-binding cassette transporter ABCC4 was identified as a modulator of SHH-MB. Accordingly, increased ABCC4 expression correlated with poor overall survival in SHH MB patients. Knockdown of ABCC4 expression markedly blunted the constitutive activation of the SHH pathway secondary to Ptch1 or Sufu insufficiency. In human tumor cell lines, ABCC4 knockdown and inhibition reduced full-length GLI3 levels. In a clinically relevant murine SHH MB model, targeted ablation of Abcc4 in primary tumors significantly reduced tumor burden and extended the lifespan of tumor-bearing mice. These studies reveal ABCC4 as a potent SHH pathway regulator and a new candidate to target with the potential to improve SHH MB therapy.
-
Muscle-Derived Dpp Regulates Feeding Initiation via Endocrine Modulation of Brain Dopamine BiosynthesisGenes & Development 2020
In animals, the brain regulates feeding behavior in response to local energy demands of peripheral tissues, which secrete orexigenic and anorexigenic hormones. Although skeletal muscle is a key peripheral tissue, it remains unknown whether muscle-secreted hormones regulate feeding. In Drosophila, we found that decapentaplegic (dpp), the homolog of human bone morphogenetic proteins BMP2 and BMP4, is a muscle-secreted factor (a myokine) that is induced by nutrient sensing and that circulates and signals to the brain. Muscle-restricted dpp RNAi promotes foraging and feeding initiation, whereas dpp overexpression reduces it. This regulation of feeding by muscle-derived Dpp stems from modulation of brain tyrosine hydroxylase (TH) expression and dopamine biosynthesis. Consistently, Dpp receptor signaling in dopaminergic neurons regulates TH expression and feeding initiation via the downstream transcriptional repressor Schnurri. Moreover, pharmacologic modulation of TH activity rescues the changes in feeding initiation due to modulation of dpp expression in muscle. These findings indicate that muscle-to-brain endocrine signaling mediated by the myokine Dpp regulates feeding behavior.
2019:10
-
Targeting REGNASE-1 Programs Long-Lived Effector T Cells for Cancer TherapyNature 2019
Adoptive cell therapy represents a new paradigm in cancer immunotherapy, but it can be limited by the poor persistence and function of transferred T cells1. Here we use an in vivo pooled CRISPR–Cas9 mutagenesis screening approach to demonstrate that, by targeting REGNASE-1, CD8+ T cells are reprogrammed to long-lived effector cells with extensive accumulation, better persistence and robust effector function in tumours. REGNASE-1-deficient CD8+ T cells show markedly improved therapeutic efficacy against mouse models of melanoma and leukaemia. By using a secondary genome-scale CRISPR–Cas9 screening, we identify BATF as the key target of REGNASE-1 and as a rheostat that shapes antitumour responses. Loss of BATF suppresses the increased accumulation and mitochondrial fitness of REGNASE-1-deficient CD8+ T cells. By contrast, the targeting of additional signalling factors—including PTPN2 and SOCS1—improves the therapeutic efficacy of REGNASE-1-deficient CD8+ T cells. Our findings suggest that T cell persistence and effector function can be coordinated in tumour immunity and point to avenues for improving the efficacy of adoptive cell therapy for cancer.
-
Matters of Life and Death: How Estrogen and Estrogen Receptor Binding to the Immunoglobulin Heavy Chain Locus May Influence Outcomes of Infection, Allergy, and Autoimmune DiseaseCellular Immunology 2019
Sex hormones are best known for their influences on reproduction, but they also have profound influences on the immune response. Examples of sex-specific differences include: (i) the relatively poor control of influenza virus infections in males compared to females, (ii) allergic asthma, an IgE-associated hypersensitivity reaction that is exacerbated in adolescent females compared to males, and (iii) systemic lupus erythematosus, a life-threatening autoimmune disease with a 9:1 female:male bias. Here we consider how estrogen and estrogen receptor α (ERα) may influence the immune response by modifying class switch recombination (CSR) and immunoglobulin expression patterns. We focus on ERα binding to enhancers (Eμ and the 3’ regulatory region) and switch sites (S\textmu and Sε) in the immunoglobulin heavy chain locus. Our preliminary data from ChIP-seq analyses of purified, activated B cells show estrogen-mediated changes in the positioning of ERα binding within and near S\textmu and Sε. In the presence of estrogen, ERα is bound not only to estrogen response elements (ERE), but also to adenosine-cytidine (AC)-repeats and poly adenosine (poly A) sequences, in some cases within constant region gene introns. We propose that by binding these sites, estrogen and ERα directly participate in the DNA loop formation required for CSR. We further suggest that estrogen regulates immunoglobulin expression patterns and can thereby influence life-and-death outcomes of infection, hypersensitivity, and autoimmune disease.
-
Nucleome Dynamics during Retinal DevelopmentNeuron 2019
Summary More than 8,000 genes are turned on or off as progenitor cells produce the 7 classes of retinal cell types during development. Thousands of enhancers are also active in the developing retinae, many having features of cell- and developmental stage-specific activity. We studied dynamic changes in the 3D chromatin landscape important for precisely orchestrated changes in gene expression during retinal development by ultra-deep in situ Hi-C analysis on murine retinae. We identified developmental-stage-specific changes in chromatin compartments and enhancer-promoter interactions. We developed a machine learning-based algorithm to map euchromatin and heterochromatin domains genome-wide and overlaid it with chromatin compartments identified by Hi-C. Single-cell ATAC-seq and RNA-seq were integrated with our Hi-C and previous ChIP-seq data to identify cell- and developmental-stage-specific super-enhancers (SEs). We identified a bipolar neuron-specific core regulatory circuit SE upstream of Vsx2, whose deletion in mice led to the loss of bipolar neurons.
-
Acute Depletion of CTCF Directly Affects MYC Regulation through Loss of Enhancer–Promoter LoopingNucleic Acids Research 2019
Numerous pieces of evidence support the complex, 3D spatial organization of the genome dictates gene expression. CTCF is essential to define topologically associated domain boundaries and to facilitate the formation of insulated chromatin loop structures. To understand CTCF’s direct role in global transcriptional regulation, we integrated the miniAID-mClover3 cassette to the endogenous CTCF locus in a human pediatric B-ALL cell line, SEM, and an immortal erythroid precursor cell line, HUDEP-2, to allow for acute depletion of CTCF protein by the auxin-inducible degron system. In SEM cells, CTCF loss notably disrupted intra-TAD loops and TAD integrity in concurrence with a reduction in CTCF-binding affinity, while showing no perturbation to nuclear compartment integrity. Strikingly, the overall effect of CTCF’s loss on transcription was minimal. Whole transcriptome analysis showed hundreds of genes differentially expressed in CTCF-depleted cells, among which MYC and a number of MYC target genes were specifically downregulated. Mechanically, acute depletion of CTCF disrupted the direct interaction between the MYC promoter and its distal enhancer cluster residing ∼1.8 Mb downstream. Notably, MYC expression was not profoundly affected upon CTCF loss in HUDEP-2 cells suggesting that CTCF could play a B-ALL cell line specific role in maintaining MYC expression.
-
Long-Read Sequencing Unveils IGH-DUX4 Translocation into the Silenced IGH Allele in B-Cell Acute Lymphoblastic LeukemiaNature Communications 2019
IGH@ proto-oncogene translocation is a common oncogenic event in lymphoid lineage cancers such as B-ALL, lymphoma and multiple myeloma. Here, to investigate the interplay between IGH@ proto-oncogene translocation and IGH allelic exclusion, we perform long-read whole-genome and transcriptome sequencing along with epigenetic and 3D genome profiling of Nalm6, an IGH-DUX4 positive B-ALL cell line. We detect significant allelic imbalance on the wild-type over the IGH-DUX4 haplotype in expression and epigenetic data, showing IGH-DUX4 translocation occurs on the silenced IGH allele. In vitro, this reduces the oncogenic stress of DUX4 high-level expression. Moreover, patient samples of IGH-DUX4 B-ALL have similar expression profile and IGH breakpoints as Nalm6, suggesting a common mechanism to allow optimal dosage of non-toxic DUX4 expression.
-
H3.3 K27M Depletion Increases Differentiation and Extends Latency of Diffuse Intrinsic Pontine Glioma Growth in VivoActa Neuropathologica 2019
Histone H3 K27M mutation is the defining molecular feature of the devastating pediatric brain tumor, diffuse intrinsic pontine glioma (DIPG). The prevalence of histone H3 K27M mutations indicates a critical role in DIPGs, but the contribution of the mutation to disease pathogenesis remains unclear. We show that knockdown of this mutation in DIPG xenografts restores K27M-dependent loss of H3K27me3 and delays tumor growth. Comparisons of matched DIPG xenografts with and without K27M knockdown allowed identification of mutation-specific effects on the transcriptome and epigenome. The resulting transcriptional changes recapitulate expression signatures from K27M primary DIPG tumors and are strongly enriched for genes associated with nervous system development. Integrated analysis of ChIP-seq and expression data showed that genes upregulated by the mutation are overrepresented in apparently bivalent promoters. Many of these targets are associated with more immature differentiation states. Expression profiles indicate K27M knockdown decreases proliferation and increases differentiation within lineages represented in DIPG. These data suggest that K27M-mediated loss of H3K27me3 directly regulates a subset of genes by releasing poised promoters, and contributes to tumor phenotype and growth by limiting differentiation. The delayed tumor growth associated with knockdown of H3 K27M provides evidence that this highly recurrent mutation is a relevant therapeutic target.
-
Complex Sex-Biased Antibody Responses: Estrogen Receptors Bind Estrogen Response Elements Centered within Immunoglobulin Heavy Chain Gene EnhancersInternational Immunology 2019
Nuclear hormone receptors including the estrogen receptor (ERα) and the retinoic acid receptor regulate a plethora of biological functions including reproduction, circulation and immunity. To understand how estrogen and other nuclear hormones influence antibody production, we characterized total serum antibody isotypes in female and male mice of C57BL/6J, BALB/cJ and C3H/HeJ mouse strains. Antibody levels were higher in females compared to males in all strains and there was a female preference for IgG2b production. Sex-biased patterns were influenced by vitamin levels, and by antigen specificity toward influenza virus or pneumococcus antigens. To help explain sex biases, we examined the direct effects of estrogen on immunoglobulin heavy chain sterile transcript production among purified, lipopolysaccharide-stimulated B cells. Supplemental estrogen in B-cell cultures significantly increased immunoglobulin heavy chain sterile transcripts. Chromatin immunoprecipitation analyses of activated B cells identified significant ERα binding to estrogen response elements (EREs) centered within enhancer elements of the immunoglobulin heavy chain locus, including the E\textmu enhancer and hypersensitive site 1,2 (HS1,2) in the 3’ regulatory region. The ERE in HS1,2 was conserved across animal species, and in humans marked a site of polymorphism associated with the estrogen-augmented autoimmune disease, lupus. Taken together, the results highlight: (i) the important targets of ERα in regulatory regions of the immunoglobulin heavy chain locus that influence antibody production, and (ii) the complexity of mechanisms by which estrogen instructs sex-biased antibody production profiles.
-
Differentiation of Human Pluripotent Stem Cells into Neurons or Cortical Organoids Requires Transcriptional Co-Regulation by UTX and 53BP1Nature Neuroscience 2019
UTX is a chromatin modifier required for development and neural lineage specification, but how it controls these biological processes is unclear. To determine the molecular mechanisms of UTX, we identified novel UTX protein interaction partners. Here we show that UTX and 53BP1 directly interact and co-occupy promoters in human embryonic stem cells and differentiating neural progenitor cells. Human 53BP1 contains a UTX-binding site that diverges from its mouse homolog by 41%, and disruption of the 53BP1–UTX interaction abrogated human, but not mouse, neurogenesis in vitro. The 53BP1–UTX interaction is required to upregulate key neurodevelopmental genes during the differentiation of human embryonic stem cells into neurons or into cortical organoids. 53BP1 promotes UTX chromatin binding, and in turn H3K27 modifications and gene activation, at a subset of genomic regions, including neurogenic genes. Overall, our data suggest that the 53BP1–UTX interaction supports the activation of key genes required for human neurodevelopment.
-
Metabolic Heterogeneity Underlies Reciprocal Fates of T H 17 Cell Stemness and PlasticityNature 2019
A defining feature of adaptive immunity is the development of long-lived memory T cells to curtail infection. Recent studies have identified a unique stem-like T-cell subset amongst exhausted CD8-positive T cells in chronic infection1–3, but it remains unclear whether CD4-positive T-cell subsets with similar features exist in chronic inflammatory conditions. Amongst helper T cells, TH17 cells have prominent roles in autoimmunity and tissue inflammation and are characterized by inherent plasticity4–7, although how such plasticity is regulated is poorly understood. Here we demonstrate that TH17 cells in a mouse model of autoimmune disease are functionally and metabolically heterogeneous; they contain a subset with stemness-associated features but lower anabolic metabolism, and a reciprocal subset with higher metabolic activity that supports transdifferentiation into TH1-like cells. These two TH17-cell subsets are defined by selective expression of the transcription factors TCF-1 and T-bet, and by discrete levels of CD27 expression. We also identify signalling via the kinase complex mTORC1 as a central regulator of TH17-cell fate decisions by coordinating metabolic and transcriptional programmes. TH17 cells with disrupted mTORC1 signalling or anabolic metabolism fail to induce autoimmune neuroinflammation or to develop into TH1-like cells, but instead upregulate TCF-1 expression and acquire stemness-associated features. Single-cell RNA sequencing and experimental validation reveal heterogeneity in fate-mapped TH17 cells, and a developmental arrest in the TH1 transdifferentiation trajectory upon loss of mTORC1 activity or metabolic perturbation. Our results establish that the dichotomy of stemness and effector function underlies the heterogeneous TH17 responses and autoimmune pathogenesis, and point to previously unappreciated metabolic control of plasticity in helper T cells.
-
Histone H3.3 K27M Accelerates Spontaneous Brainstem Glioma and Drives Restricted Changes in Bivalent Gene ExpressionCancer Cell 2019
Summary Diffuse intrinsic pontine gliomas (DIPGs) are incurable childhood brainstem tumors with frequent histone H3 K27M mutations and recurrent alterations in PDGFRA and TP53. We generated genetically engineered inducible mice and showed that H3.3 K27M enhanced neural stem cell self-renewal while preserving regional identity. Neonatal induction of H3.3 K27M cooperated with activating platelet-derived growth factor receptor α (PDGFRα) mutant and Trp53 loss to accelerate development of diffuse brainstem gliomas that recapitulated human DIPG gene expression signatures and showed global changes in H3K27 posttranslational modifications, but relatively restricted gene expression changes. Genes upregulated in H3.3 K27M tumors were enriched for those associated with neural development where H3K27me3 loss released the poised state of apparently bivalent promoters, whereas downregulated genes were enriched for those encoding homeodomain transcription factors.
2018:6
-
The Hippo Pathway Prevents YAP/TAZ-Driven Hypertranscription and Controls Neural Progenitor NumberDevelopmental Cell 2018
Summary The Hippo pathway controls the activity of YAP/TAZ transcriptional coactivators through a kinase cascade. Despite the critical role of this pathway in tissue growth and tumorigenesis, it remains unclear how YAP/TAZ-mediated transcription drives proliferation. By analyzing the effects of inactivating LATS1/2 kinases, the direct upstream inhibitors of YAP/TAZ, on mouse brain development and applying cell-number-normalized transcriptome analyses, we discovered that YAP/TAZ activation causes a global increase in transcription activity, known as hypertranscription, and upregulates many genes associated with cell growth and proliferation. In contrast, conventional read-depth-normalized RNA-sequencing analysis failed to detect the scope of the transcriptome shift and missed most relevant gene ontologies. Following a transient increase in proliferation, however, hypertranscription in neural progenitors triggers replication stress, DNA damage, and p53 activation, resulting in massive apoptosis. Our findings reveal a significant impact of YAP/TAZ activation on global transcription activity and have important implications for understanding YAP/TAZ function.
-
The Genetic Basis and Cell of Origin of Mixed Phenotype Acute LeukaemiaNature 2018
Mixed phenotype acute leukaemia (MPAL) is a high-risk subtype of leukaemia with myeloid and lymphoid features, limited genetic characterization, and a lack of consensus regarding appropriate therapy. Here we show that the two principal subtypes of MPAL, T/myeloid (T/M) and B/myeloid (B/M), are genetically distinct. Rearrangement of ZNF384 is common in B/M MPAL, and biallelic WT1 alterations are common in T/M MPAL, which shares genomic features with early T-cell precursor acute lymphoblastic leukaemia. We show that the intratumoral immunophenotypic heterogeneity characteristic of MPAL is independent of somatic genetic variation, that founding lesions arise in primitive haematopoietic progenitors, and that individual phenotypic subpopulations can reconstitute the immunophenotypic diversity in vivo. These findings indicate that the cell of origin and founding lesions, rather than an accumulation of distinct genomic alterations, prime tumour cells for lineage promiscuity. Moreover, these findings position MPAL in the spectrum of immature leukaemias and provide a genetically informed framework for future clinical trials of potential treatments for MPAL.
-
Identification of Therapeutic Targets in Rhabdomyosarcoma through Integrated Genomic, Epigenomic, and Proteomic AnalysesCancer Cell 2018
Summary Personalized cancer therapy targeting somatic mutations in patient tumors is increasingly being incorporated into practice. Other therapeutic vulnerabilities resulting from changes in gene expression due to tumor specific epigenetic perturbations are progressively being recognized. These genomic and epigenomic changes are ultimately manifest in the tumor proteome and phosphoproteome. We integrated transcriptomic, epigenomic, and proteomic/phosphoproteomic data to elucidate the cellular origins and therapeutic vulnerabilities of rhabdomyosarcoma (RMS). We discovered that alveolar RMS occurs further along the developmental program than embryonal RMS. We also identified deregulation of the RAS/MEK/ERK/CDK4/6, G2/M, and unfolded protein response pathways through our integrated analysis. Comprehensive preclinical testing revealed that targeting the WEE1 kinase in the G2/M pathway is the most effective approach in vivo for high-risk RMS.
-
Mouse Medulloblastoma Driven by CRISPR Activation of Cellular MycScientific Reports 2018
MYC-driven Group 3 (G3) medulloblastoma (MB) is the most aggressive of four molecular subgroups classified by transcriptome, genomic landscape and clinical outcomes. Mouse models that recapitulate human G3 MB all rely on retroviral vector-induced Myc expression driven by viral regulatory elements (Retro-Myc tumors). We used nuclease-deficient CRISPR/dCas9-based gene activation with combinatorial single guide RNAs (sgRNAs) to enforce transcription of endogenous Myc in Trp53-null neurospheres that were orthotopically transplanted into the brains of naïve animals. Three combined sgRNAs linked to dCas9-VP160 induced cellular Myc expression and large cell anaplastic MBs (CRISPR-Myc tumors) which recapitulated the molecular characteristics of mouse and human G3 MBs. The BET inhibitor JQ1 suppressed MYC expression in a human G3 MB cell line (HD-MB03) and CRISPR-Myc, but not in Retro-Myc MBs. This G3 MB mouse model in which Myc expression is regulated by its own promoter will facilitate pre-clinical studies with drugs that regulate Myc transcription.
-
MYC Drives a Subset of High-Risk Pediatric Neuroblastomas and Is Activated through Mechanisms Including Enhancer Hijacking and Focal Enhancer AmplificationCancer Discovery 2018
The amplified MYCN gene serves as an oncogenic driver in approximately 20% of high-risk pediatric neuroblastomas. Here, we show that the family member MYC is a potent transforming gene in a separate subset of high-risk neuroblastoma cases (∼10%), based on (i) its upregulation by focal enhancer amplification or genomic rearrangements leading to enhancer hijacking, and (ii) its ability to transform neuroblastoma precursor cells in a transgenic animal model. The aberrant regulatory elements associated with oncogenic MYC activation include focally amplified distal enhancers and translocation of highly active enhancers from other genes to within topologically associating domains containing the MYC gene locus. The clinical outcome for patients with high levels of MYC expression is virtually identical to that of patients with amplification of the MYCN gene, a known high-risk feature of this disease. Together, these findings establish MYC as a bona fide oncogene in a clinically significant group of high-risk childhood neuroblastomas. Significance: Amplification of the MYCN oncogene is a recognized hallmark of high-risk pediatric neuroblastoma. Here, we demonstrate that MYC is also activated as a potent oncogene in a distinct subset of neuroblastoma cases through either focal amplification of distal enhancers or enhancer hijacking mediated by chromosomal translocation. Cancer Discov; 8(3); 320–35. \textcopyright 2017 AACR. This article is highlighted in the In This Issue feature, p. 253
-
Retinal Cell Type DNA Methylation and Histone Modifications Predict Reprogramming Efficiency and Retinogenesis in 3D Organoid CulturesCell Reports 2018
Summary Diverse cell types can be reprogrammed into pluripotent stem cells by ectopic expression of Oct4 (Pou5f1), Klf4, Sox2, and Myc. Many of these induced pluripotent stem cells (iPSCs) retain memory, in terms of DNA methylation and histone modifications (epigenetic memory), of their cellular origins, and this may bias subsequent differentiation. Neurons are difficult to reprogram, and there has not been a systematic side-by-side characterization of reprogramming efficiency or epigenetic memory across different neuronal subtypes. Here, we compare reprogramming efficiency of five different retinal cell types at two different stages of development. Retinal differentiation from each iPSC line was measured using a quantitative standardized scoring system called STEM-RET and compared to the epigenetic memory. Neurons with the lowest reprogramming efficiency produced iPSC lines with the best retinal differentiation and were more likely to retain epigenetic memory of their cellular origins. In addition, we identified biomarkers of iPSCs that are predictive of retinal differentiation.
2017:2
-
Orthotopic Patient-Derived Xenografts of Paediatric Solid TumoursNature 2017
Preclinical models of paediatric solid tumours that could help identify predictive biomarkers of a patient’s sensitivity to therapy have been lacking. Over five years, the authors have developed an open access collection of orthotopic xenografts of 12 types of paediatric tumour. Genomic and epigenetic characterization reveals that xenografts retain characteristics of the tumour of origin. A high-throughput drug screen provides a resource for the community to identify potentially efficacious drug combinations.
-
The Dynamic Epigenetic Landscape of the Retina During Development, Reprogramming, and TumorigenesisNeuron 2017
In the developing retina, multipotent neural progenitors undergo unidirectional differentiation in a precise spatiotemporal order. Here we profile the epigenetic and transcriptional changes that occur during retinogenesis in mice and humans. Although some progenitor genes and cell cycle genes were epigenetically silenced during retinogenesis, the most dramatic change was derepression of cell-type-specific differentiation programs. We identified developmental-stage-specific super-enhancers and showed that most epigenetic changes are conserved in humans and mice. To determine how the epigenome changes during tumorigenesis and reprogramming, we performed integrated epigenetic analysis of murine and human retinoblastomas and induced pluripotent stem cells (iPSCs) derived from murine rod photoreceptors. The retinoblastoma epigenome mapped to the developmental stage when retinal progenitors switch from neurogenic to terminal patterns of cell division. The epigenome of retinoblastomas was more similar to that of the normal retina than that of retina-derived iPSCs, and we identified retina-specific epigenetic memory.
2016:3
-
Deregulation of DUX4 and ERG in Acute Lymphoblastic LeukemiaNature Genetics 2016
Chromosomal rearrangements deregulating hematopoietic transcription factors are common in acute lymphoblastic leukemia (ALL). Here we show that deregulation of the homeobox transcription factor gene DUX4 and the ETS transcription factor gene ERG is a hallmark of a subtype of B-progenitor ALL that comprises up to 7% of B-ALL. DUX4 rearrangement and overexpression was present in all cases and was accompanied by transcriptional deregulation of ERG, expression of a novel ERG isoform, ERGalt, and frequent ERG deletion. ERGalt uses a non-canonical first exon whose transcription was initiated by DUX4 binding. ERGalt retains the DNA-binding and transactivation domains of ERG, but it inhibits wild-type ERG transcriptional activity and is transforming. These results illustrate a unique paradigm of transcription factor deregulation in leukemia in which DUX4 deregulation results in loss of function of ERG, either by deletion or induced expression of an isoform that is a dominant-negative inhibitor of wild-type ERG function.
-
Binding of Estrogen Receptors to Switch Sites and Regulatory Elements in the Immunoglobulin Heavy Chain Locus of Activated B Cells Suggests a Direct Influence of Estrogen on Antibody ExpressionMolecular Immunology 2016
Females and males differ in antibody isotype expression patterns and in immune responses to foreign- and self-antigens. For example, systemic lupus erythematosus is a condition that associates with the production of isotype-skewed anti-self antibodies, and exhibits a 9:1 female:male disease ratio. To explain differences between B cell responses in males and females, we sought to identify direct interactions of the estrogen receptor (ER) with the immunoglobulin heavy chain locus. This effort was encouraged by our previous identification of estrogen response elements (ERE) in heavy chain switch (S) regions. We conducted a full-genome chromatin immunoprecipitation analysis (ChIP-seq) using DNA from LPS-activated B cells and an ERα-specific antibody. Results revealed ER binding to a wide region of DNA, spanning sequences from the JH cluster to Cδ, with peaks in Eμ and Sμ sites. Additional peaks of ERα binding were coincident with hs1,2 and hs4 sites in the 3’ regulatory region (3’RR) of the heavy chain locus. This first demonstration of direct binding of ER to key regulatory elements in the immunoglobulin locus supports our hypothesis that estrogen and other nuclear hormone receptors and ligands may directly influence antibody expression and class switch recombination (CSR). Our hypothesis encourages the conduct of new experiments to evaluate the consequences of ER binding. A better understanding of ER:DNA interactions in the immunoglobulin heavy chain locus, and respective mechanisms, may ultimately translate to better control of antibody expression, better protection against pathogens, and prevention of pathologies caused by auto-immune disease.
-
Hotspots for Vitamin–Steroid–Thyroid Hormone Response Elements Within Switch Regions of Immunoglobulin Heavy Chain Loci Predict a Direct Influence of Vitamins and Hormones on B Cell Class Switch RecombinationViral Immunology 2016
Vitamin A deficiencies are common throughout the world and have a significant negative influence on immune protection against viral infections. Mouse models demonstrate that the production of IgA, a first line of defense against viruses at mucosal sites, is inhibited in the context of vitamin A deficiency. In vitro, the addition of vitamin A to activated B cells can enhance IgA expression, but downregulate IgE. Previous reports have demonstrated that vitamin A modifies cytokine patterns, and in so doing may influence antibody isotype expression by an indirect mechanism. However, we have now discovered hundreds of potential response elements among Sμ, S\varepsilon, and Sα switch sites within immunoglobulin heavy chain loci. These hotspots appear in both mouse and human loci and include targets for vitamin receptors and related proteins (e.g., estrogen receptors) in the nuclear receptor superfamily. Full response elements with direct repeats are relatively infrequent or absent in Sγ regions although half-sites are present. Based on these results, we pose a hypothesis that nuclear receptors have a direct effect on the immunoglobulin heavy chain class switch recombination event. We propose that vitamin A may alter S site accessibility to activation-induced deaminase and nonhomologous end-joining machinery, thereby influencing the isotype switch, antibody production, and protection against viral infections at mucosal sites.
2015:3
-
The Glucose-Sensing Transcription Factor MLX Promotes Myogenesis via Myokine SignalingGenes & Development 2015
Metabolic stress and changes in nutrient levels modulate many aspects of skeletal muscle function during aging and disease. Growth factors and cytokines secreted by skeletal muscle, known as myokines, are important signaling factors, but it is largely unknown whether they modulate muscle growth and differentiation in response to nutrients. Here, we found that changes in glucose levels increase the activity of the glucose-responsive transcription factor MLX (Max-like protein X), which promotes and is necessary for myoblast fusion. MLX promotes myogenesis not via an adjustment of glucose metabolism but rather by inducing the expression of several myokines, including insulin-like growth factor 2 (IGF2), whereas RNAi and dominant-negative MLX reduce IGF2 expression and block myogenesis. This phenotype is rescued by conditioned medium from control muscle cells and by recombinant IGF2, which activates the myogenic kinase Akt. Importantly, MLX-null mice display decreased IGF2 induction and diminished muscle regeneration in response to injury, indicating that the myogenic function of MLX is manifested in vivo. Thus, glucose is a signaling molecule that regulates myogenesis and muscle regeneration via MLX/IGF2/Akt signaling.
-
Brg1 Coordinates Multiple Processes during Retinogenesis and Is a Tumor Suppressor in RetinoblastomaDevelopment 2015
Skip to Next Section Retinal development requires precise temporal and spatial coordination of cell cycle exit, cell fate specification, cell migration and differentiation. When this process is disrupted, retinoblastoma, a developmental tumor of the retina, can form. Epigenetic modulators are central to precisely coordinating developmental events, and many epigenetic processes have been implicated in cancer. Studying epigenetic mechanisms in development is challenging because they often regulate multiple cellular processes; therefore, elucidating the primary molecular mechanisms involved can be difficult. Here we explore the role of Brg1 (Smarca4) in retinal development and retinoblastoma in mice using molecular and cellular approaches. Brg1 was found to regulate retinal size by controlling cell cycle length, cell cycle exit and cell survival during development. Brg1 was not required for cell fate specification but was required for photoreceptor differentiation and cell adhesion/polarity programs that contribute to proper retinal lamination during development. The combination of defective cell differentiation and lamination led to retinal degeneration in Brg1-deficient retinae. Despite the hypocellularity, premature cell cycle exit, increased cell death and extended cell cycle length, retinal progenitor cells persisted in Brg1-deficient retinae, making them more susceptible to retinoblastoma. ChIP-Seq analysis suggests that Brg1 might regulate gene expression through multiple mechanisms.
-
Quantification of Retinogenesis in 3D Cultures Reveals Epigenetic Memory and Higher Efficiency in iPSCs Derived from Rod PhotoreceptorsCell Stem Cell 2015
Summary Cell-based therapies to treat retinal degeneration are now being tested in clinical trials. However, it is not known whether the source of stem cells is important for the production of differentiated cells suitable for transplantation. To test this, we generated induced pluripotent stem cells (iPSCs) from murine rod photoreceptors (r-iPSCs) and scored their ability to make retinae by using a standardized quantitative protocol called STEM-RET. We discovered that r-iPSCs more efficiently produced differentiated retinae than did embryonic stem cells (ESCs) or fibroblast-derived iPSCs (f-iPSCs). Retinae derived from f-iPSCs had fewer amacrine cells and other inner nuclear layer cells. Integrated epigenetic analysis showed that DNA methylation contributes to the defects in f-iPSC retinogenesis and that rod-specific CTCF insulator protein-binding sites may promote r-iPSC retinogenesis. Together, our data suggest that the source of stem cells is important for producing retinal neurons in three-dimensional (3D) organ cultures.